当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Electrophilic Aromatic Sulfonation with SO3: Concerted or Classic SEAr Mechanism?
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2011-11-30 , DOI: 10.1021/ja201866h
Gergana Koleva 1 , Boris Galabov 1 , Jing Kong 2 , Henry F. Schaefer 2 , Paul von R. Schleyer 2
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2011-11-30 , DOI: 10.1021/ja201866h
Gergana Koleva 1 , Boris Galabov 1 , Jing Kong 2 , Henry F. Schaefer 2 , Paul von R. Schleyer 2
Affiliation
|
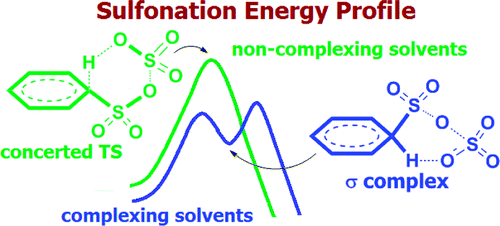
The electrophilic sulfonation of several arenes with SO(3) was examined by electronic structure computations at the M06-2X/6-311+G(2d,2p) and SCS-MP2/6-311+G(2d,2p) levels of theory. In contrast to the usual interpretations, the results provide clear evidence that in nonpolar media and in the absence of catalysts the mechanism of aromatic sulfonation with a single SO(3) is concerted and does not involve the conventionally depicted 1:1 σ complex (Wheland) intermediate. Moreover, the computed activation energy for the 1:1 process is unrealistically high; barriers for alternative 2:1 mechanisms involving attack by two SO(3) molecules are 12-20 kcal/mol lower! A direct 2:1 sulfonation mechanism, involving a single essential transition state, but no Wheland type intermediate, is preferred generally at MP2 as well as at M06-2X in isolation (gas phase) or in noncomplexing solvents (such as CCl(4) or CFCl(3)). However, in polar, higher dielectric SO(3)-complexing media, M06-2X favors an S(E)Ar mechanism for the 2:1 reaction involving a Wheland-type arene-(SO(3))(2) dimer intermediate. The reaction is slower in complexing solvents, since the association energy, e.g., with nitromethane, must be overcome. But, in accord with the experimental kinetics (second-order in SO(3)), attack by two sulfur trioxide molecules is still favored energetically over reaction with a single SO(3) in CH(3)NO(2). The theoretical results also reproduce the experimental reactivity and regioselectivity trends for benzene, toluene, and naphthalene sulfonation accurately.
中文翻译:

SO3 亲电芳香族磺化:协同或经典 SEAr 机制?
通过在 M06-2X/6-311+G(2d,2p) 和 SCS-MP2/6-311+G(2d,2p) 水平的电子结构计算,研究了几种芳烃与 SO(3) 的亲电磺化理论。与通常的解释相反,结果提供了明确的证据,即在非极性介质和没有催化剂的情况下,单一 SO(3) 的芳烃磺化机制是一致的,不涉及传统描述的 1:1 σ 复合物(Wheland ) 中间的。此外,1:1 过程的计算活化能高得不切实际;涉及两个 SO(3) 分子攻击的替代 2:1 机制的屏障低 12-20 kcal/mol!直接的 2:1 磺化机制,涉及单一的基本过渡态,但没有惠兰型中间体,通常优选在 MP2 以及 M06-2X 分离(气相)或在非络合溶剂(例如 CCl(4) 或 CFCl(3))中。然而,在极性、更高介电的 SO(3)-络合介质中,M06-2X 有利于 S(E)Ar 机制,用于涉及惠兰型芳烃-(SO(3))(2) 二聚体中间体的 2:1 反应. 在络合溶剂中反应较慢,因为必须克服缔合能,例如与硝基甲烷的结合能。但是,根据实验动力学(SO(3) 中的二阶),两个三氧化硫分子的攻击仍然比 CH(3)NO(2) 中单个 SO(3) 的反应更受青睐。理论结果还准确地再现了苯、甲苯和萘磺化的实验反应性和区域选择性趋势。更高介电的 SO(3)-络合介质,M06-2X 有利于 S(E)Ar 机制,用于涉及惠兰型芳烃-(SO(3))(2) 二聚体中间体的 2:1 反应。在络合溶剂中反应较慢,因为必须克服缔合能,例如与硝基甲烷的结合能。但是,根据实验动力学(SO(3) 中的二阶),两个三氧化硫分子的攻击仍然比 CH(3)NO(2) 中单个 SO(3) 的反应更受青睐。理论结果还准确地再现了苯、甲苯和萘磺化的实验反应性和区域选择性趋势。更高介电的 SO(3)-络合介质,M06-2X 有利于 S(E)Ar 机制,用于涉及惠兰型芳烃-(SO(3))(2) 二聚体中间体的 2:1 反应。在络合溶剂中反应较慢,因为必须克服缔合能,例如与硝基甲烷的结合能。但是,根据实验动力学(SO(3) 中的二阶),两个三氧化硫分子的攻击仍然比 CH(3)NO(2) 中单个 SO(3) 的反应更受青睐。理论结果还准确地再现了苯、甲苯和萘磺化的实验反应性和区域选择性趋势。g.,与硝基甲烷,必须克服。但是,根据实验动力学(SO(3) 中的二阶),两个三氧化硫分子的攻击仍然比 CH(3)NO(2) 中单个 SO(3) 的反应更受青睐。理论结果还准确地再现了苯、甲苯和萘磺化的实验反应性和区域选择性趋势。g.,与硝基甲烷,必须克服。但是,根据实验动力学(SO(3) 中的二阶),两个三氧化硫分子的攻击仍然比 CH(3)NO(2) 中单个 SO(3) 的反应更受青睐。理论结果还准确地再现了苯、甲苯和萘磺化的实验反应性和区域选择性趋势。
更新日期:2011-11-30
中文翻译:
SO3 亲电芳香族磺化:协同或经典 SEAr 机制?
通过在 M06-2X/6-311+G(2d,2p) 和 SCS-MP2/6-311+G(2d,2p) 水平的电子结构计算,研究了几种芳烃与 SO(3) 的亲电磺化理论。与通常的解释相反,结果提供了明确的证据,即在非极性介质和没有催化剂的情况下,单一 SO(3) 的芳烃磺化机制是一致的,不涉及传统描述的 1:1 σ 复合物(Wheland ) 中间的。此外,1:1 过程的计算活化能高得不切实际;涉及两个 SO(3) 分子攻击的替代 2:1 机制的屏障低 12-20 kcal/mol!直接的 2:1 磺化机制,涉及单一的基本过渡态,但没有惠兰型中间体,通常优选在 MP2 以及 M06-2X 分离(气相)或在非络合溶剂(例如 CCl(4) 或 CFCl(3))中。然而,在极性、更高介电的 SO(3)-络合介质中,M06-2X 有利于 S(E)Ar 机制,用于涉及惠兰型芳烃-(SO(3))(2) 二聚体中间体的 2:1 反应. 在络合溶剂中反应较慢,因为必须克服缔合能,例如与硝基甲烷的结合能。但是,根据实验动力学(SO(3) 中的二阶),两个三氧化硫分子的攻击仍然比 CH(3)NO(2) 中单个 SO(3) 的反应更受青睐。理论结果还准确地再现了苯、甲苯和萘磺化的实验反应性和区域选择性趋势。更高介电的 SO(3)-络合介质,M06-2X 有利于 S(E)Ar 机制,用于涉及惠兰型芳烃-(SO(3))(2) 二聚体中间体的 2:1 反应。在络合溶剂中反应较慢,因为必须克服缔合能,例如与硝基甲烷的结合能。但是,根据实验动力学(SO(3) 中的二阶),两个三氧化硫分子的攻击仍然比 CH(3)NO(2) 中单个 SO(3) 的反应更受青睐。理论结果还准确地再现了苯、甲苯和萘磺化的实验反应性和区域选择性趋势。更高介电的 SO(3)-络合介质,M06-2X 有利于 S(E)Ar 机制,用于涉及惠兰型芳烃-(SO(3))(2) 二聚体中间体的 2:1 反应。在络合溶剂中反应较慢,因为必须克服缔合能,例如与硝基甲烷的结合能。但是,根据实验动力学(SO(3) 中的二阶),两个三氧化硫分子的攻击仍然比 CH(3)NO(2) 中单个 SO(3) 的反应更受青睐。理论结果还准确地再现了苯、甲苯和萘磺化的实验反应性和区域选择性趋势。g.,与硝基甲烷,必须克服。但是,根据实验动力学(SO(3) 中的二阶),两个三氧化硫分子的攻击仍然比 CH(3)NO(2) 中单个 SO(3) 的反应更受青睐。理论结果还准确地再现了苯、甲苯和萘磺化的实验反应性和区域选择性趋势。g.,与硝基甲烷,必须克服。但是,根据实验动力学(SO(3) 中的二阶),两个三氧化硫分子的攻击仍然比 CH(3)NO(2) 中单个 SO(3) 的反应更受青睐。理论结果还准确地再现了苯、甲苯和萘磺化的实验反应性和区域选择性趋势。
































 京公网安备 11010802027423号
京公网安备 11010802027423号