当前位置:
X-MOL 学术
›
Adv. Energy Mater.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Rational Design of Graphene‐Supported Single Atom Catalysts for Hydrogen Evolution Reaction
Advanced Energy Materials ( IF 24.4 ) Pub Date : 2019-01-25 , DOI: 10.1002/aenm.201803689 Md Delowar Hossain 1 , Zhenjing Liu 1 , Minghao Zhuang 1 , Xingxu Yan 2 , Gui-Liang Xu 3 , Chaitanya Avinash Gadre 4 , Abhishek Tyagi 1 , Irfan Haider Abidi 1 , Cheng-Jun Sun 5 , Hoilun Wong 1 , Alexander Guda 6 , Yufeng Hao 7 , Xiaoqing Pan 2, 4 , Khalil Amine 3 , Zhengtang Luo 1
Advanced Energy Materials ( IF 24.4 ) Pub Date : 2019-01-25 , DOI: 10.1002/aenm.201803689 Md Delowar Hossain 1 , Zhenjing Liu 1 , Minghao Zhuang 1 , Xingxu Yan 2 , Gui-Liang Xu 3 , Chaitanya Avinash Gadre 4 , Abhishek Tyagi 1 , Irfan Haider Abidi 1 , Cheng-Jun Sun 5 , Hoilun Wong 1 , Alexander Guda 6 , Yufeng Hao 7 , Xiaoqing Pan 2, 4 , Khalil Amine 3 , Zhengtang Luo 1
Affiliation
|
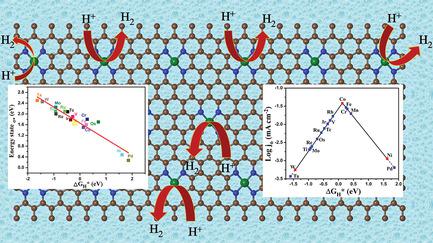
The proper choice of nonprecious transition metals as single atom catalysts (SACs) remains unclear for designing highly efficient electrocatalysts for hydrogen evolution reaction (HER). Herein, reported is an activity correlation with catalysts, electronic structure, in order to clarify the origin of reactivity for a series of transition metals supported on nitrogen‐doped graphene as SACs for HER by a combination of density functional theory calculations and electrochemical measurements. Only few of the transition metals (e.g., Co, Cr, Fe, Rh, and V) as SACs show good catalytic activity toward HER as their Gibbs free energies are varied between the range of –0.20 to 0.30 eV but among which Co‐SAC exhibits the highest electrochemical activity at 0.13 eV. Electronic structure studies show that the energy states of active valence dz2 orbitals and their resulting antibonding state determine the catalytic activity for HER. The fact that the antibonding state orbital is neither completely empty nor fully filled in the case of Co‐SAC is the main reason for its ideal hydrogen adsorption energy. Moreover, the electrochemical measurement shows that Co‐SAC exhibits a superior hydrogen evolution activity over Ni‐SAC and W‐SAC, confirming the theoretical calculation. This systematic study gives a fundamental understanding about the design of highly efficient SACs for HER.
中文翻译:

石墨烯支持的单原子催化氢发生反应的催化剂的合理设计
对于设计用于析氢反应(HER)的高效电催化剂,尚不清楚选择非贵金属作为单原子催化剂(SAC)的过渡金属。此处,报道了与催化剂,电子结构的活性相关性,目的是通过结合密度泛函理论计算和电化学测量来阐明一系列负载在氮掺杂石墨烯上的过渡金属作为HER的SAC的反应性起源。作为SAC的过渡金属(例如Co,Cr,Fe,Rh和V)中只有极少数对HER表现出良好的催化活性,因为它们的吉布斯自由能在–0.20至0.30 eV的范围内变化,但其中Co-SAC在0.13 eV时具有最高的电化学活性。电子结构研究表明,活性价d的能态z 2轨道及其产生的反键状态决定了HER的催化活性。在Co-SAC情况下,反键态轨道既不会完全为空也不会完全填充,这是其理想的氢吸附能的主要原因。此外,电化学测量表明,Co-SAC的析氢活性优于Ni-SAC和W-SAC,这证明了理论计算的正确性。这项系统的研究对HER的高效SAC的设计提供了基本的了解。
更新日期:2019-01-25
中文翻译:
石墨烯支持的单原子催化氢发生反应的催化剂的合理设计
对于设计用于析氢反应(HER)的高效电催化剂,尚不清楚选择非贵金属作为单原子催化剂(SAC)的过渡金属。此处,报道了与催化剂,电子结构的活性相关性,目的是通过结合密度泛函理论计算和电化学测量来阐明一系列负载在氮掺杂石墨烯上的过渡金属作为HER的SAC的反应性起源。作为SAC的过渡金属(例如Co,Cr,Fe,Rh和V)中只有极少数对HER表现出良好的催化活性,因为它们的吉布斯自由能在–0.20至0.30 eV的范围内变化,但其中Co-SAC在0.13 eV时具有最高的电化学活性。电子结构研究表明,活性价d的能态z 2轨道及其产生的反键状态决定了HER的催化活性。在Co-SAC情况下,反键态轨道既不会完全为空也不会完全填充,这是其理想的氢吸附能的主要原因。此外,电化学测量表明,Co-SAC的析氢活性优于Ni-SAC和W-SAC,这证明了理论计算的正确性。这项系统的研究对HER的高效SAC的设计提供了基本的了解。
































 京公网安备 11010802027423号
京公网安备 11010802027423号