当前位置:
X-MOL 学术
›
Inorg. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Solution and Solid State Properties for Low-Spin Cobalt(II) Dibenzotetramethyltetraaza[14]annulene [(tmtaa)CoII] and the Monopyridine Complex
Inorganic Chemistry ( IF 4.3 ) Pub Date : 2019-01-08 00:00:00 , DOI: 10.1021/acs.inorgchem.8b02644 Soumyajit Dey 1 , Bradford B. Wayland 1 , Michael J. Zdilla 1
Inorganic Chemistry ( IF 4.3 ) Pub Date : 2019-01-08 00:00:00 , DOI: 10.1021/acs.inorgchem.8b02644 Soumyajit Dey 1 , Bradford B. Wayland 1 , Michael J. Zdilla 1
Affiliation
|
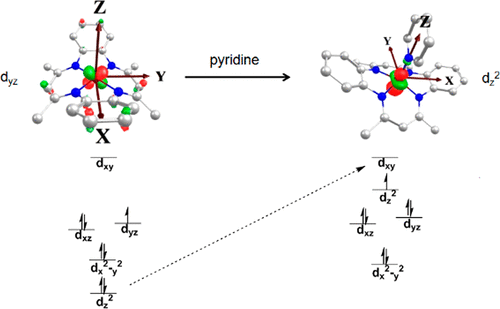
The single-crystal X-ray structure of solvent-free (tmtaa)CoII reveals three different π–π intermacrocyclic interactions between tmtaa units (tmtaa = dibenzotetramethyltetraaza[14]annulene). Pairs of inequivalent (tmtaa)CoII units in the unit cell link into a one-dimensional π–π stacked array in the solid state. Magnetic susceptibility (χ) studies from 300 to 2 K reveal the effects of intermolecular interactions between (tmtaa)CoII units in the solid state. The effective magnetic moment per CoII center is constant at 2.83 μB from 300 to 100 K and begins to significantly decrease at lower temperatures. The magnetic data are fit to a singlet (S = 0) ground state with a triplet (S = 1) excited state that is 13 cm–1 higher in energy (−2J = 13 cm–1). Toluene solutions of (tmtaa)CoII have 1H nuclear magnetic resonance (NMR) paramagnetic shifts, a solution-phase magnetic moment μeff (295 K) of 2.1 μB, and toluene glass electron paramagnetic resonance spectra that are most consistent with a low-spin (S = 1/2) CoII with the unpaired electron located in the dyz orbital. Pyridine interacts with (tmtaa)CoII to form a five-coordinate monopyridine complex in which the unpaired electron is in the dz2 orbital. The five-coordinate complex has been structurally characterized by single-crystal X-ray diffraction, and the equilibrium constant for pyridine binding at 295 K has been evaluated by both electronic and 1H NMR spectra. Density functional theory computation using the UB3LYP hybrid functional places the unpaired electron for (tmtaa)CoII in the dyz orbital and that for the monopyridine complex in the dz2 orbital, consistent with spectroscopic observations.
中文翻译:

低自旋钴(II)二苯并四甲基四氮杂[14]环戊烯[(tmtaa)Co II ]和一吡啶配合物的溶液和固态性质
无溶剂(tmtaa)Co II的单晶X射线结构揭示了tmtaa单元(tmtaa =二苯并四甲基四氮杂[14]环戊烯)之间的三种不同的π-π大环间相互作用。晶胞中成对的不等价(tmtaa)Co II单元对链接成固态的一维π-π堆叠阵列。从300到2 K的磁化率(χ)研究揭示了固态的(tmtaa)Co II单元之间的分子间相互作用的影响。每钴的有效磁矩II中心在2.83μ恒定乙从300至100 K和开始在较低温度下显著降低。磁数据拟合为单峰(小号= O)基态与三线态(S = 1)激发态,其能量高13 cm –1(-2 J = 13 cm –1)。(tmtaa)有限公司的甲苯溶液II具有1和1 H核磁共振(NMR)的顺磁位移,溶液相磁矩μ EFF 2.1μ的(295 K)乙,和甲苯玻璃电子顺磁共振光谱是与最一致低自旋(小号= 1 / 2)钴II与位于d不成对电子的yz轨道。吡啶与(tmtaa)Co II相互作用形成五配位的单吡啶配合物,其中不成对的电子在d z 2轨道上。五坐标配合物已通过单晶X射线衍射进行了结构表征,并已通过电子和1 H NMR光谱评估了295 K处吡啶结合的平衡常数。使用UB3LYP杂合函数的密度泛函理论计算将(tmtaa)Co II的不成对电子置于d yz轨道,并将单吡啶络合物的不成对电子置于d z 2轨道,与光谱观察一致。
更新日期:2019-01-08
中文翻译:
低自旋钴(II)二苯并四甲基四氮杂[14]环戊烯[(tmtaa)Co II ]和一吡啶配合物的溶液和固态性质
无溶剂(tmtaa)Co II的单晶X射线结构揭示了tmtaa单元(tmtaa =二苯并四甲基四氮杂[14]环戊烯)之间的三种不同的π-π大环间相互作用。晶胞中成对的不等价(tmtaa)Co II单元对链接成固态的一维π-π堆叠阵列。从300到2 K的磁化率(χ)研究揭示了固态的(tmtaa)Co II单元之间的分子间相互作用的影响。每钴的有效磁矩II中心在2.83μ恒定乙从300至100 K和开始在较低温度下显著降低。磁数据拟合为单峰(小号= O)基态与三线态(S = 1)激发态,其能量高13 cm –1(-2 J = 13 cm –1)。(tmtaa)有限公司的甲苯溶液II具有1和1 H核磁共振(NMR)的顺磁位移,溶液相磁矩μ EFF 2.1μ的(295 K)乙,和甲苯玻璃电子顺磁共振光谱是与最一致低自旋(小号= 1 / 2)钴II与位于d不成对电子的yz轨道。吡啶与(tmtaa)Co II相互作用形成五配位的单吡啶配合物,其中不成对的电子在d z 2轨道上。五坐标配合物已通过单晶X射线衍射进行了结构表征,并已通过电子和1 H NMR光谱评估了295 K处吡啶结合的平衡常数。使用UB3LYP杂合函数的密度泛函理论计算将(tmtaa)Co II的不成对电子置于d yz轨道,并将单吡啶络合物的不成对电子置于d z 2轨道,与光谱观察一致。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号