当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
DFT Research on Benzothiophene Pyrolysis Reaction Mechanism
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2019-01-02 00:00:00 , DOI: 10.1021/acs.jpca.8b09882
Tianshuang Li 1 , Jie Li 1 , Hongliang Zhang 1 , Kena Sun 1 , Jin Xiao 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2019-01-02 00:00:00 , DOI: 10.1021/acs.jpca.8b09882
Tianshuang Li 1 , Jie Li 1 , Hongliang Zhang 1 , Kena Sun 1 , Jin Xiao 1
Affiliation
|
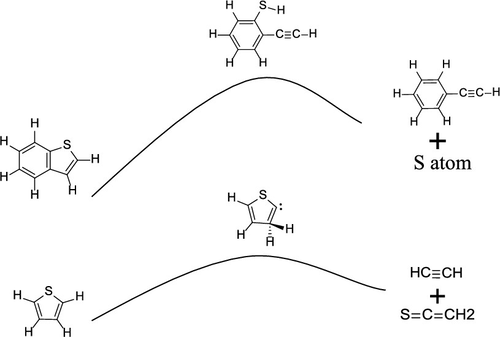
Thiophene sulfur is the most stable organic sulfur species in petroleum coke, among which benzothiophene accounts for a significant portion. Removal of benzothiophene will help to gain ultralow desulfurization. In this work, a density function theory (DFT) method was adopted to investigate benzothiophene pyrolysis mechanism. It was found that the most possible pyrolysis reaction of benzothiophene is triggered by α-H migration to β-position. The dominating products are S radical and ethenethione, which could explain benzothiophene pyrolysis experiments well. Converting thiophene fused on aromatic to a thiol group could help to promote desulfurization. As a contrast, the thiophene pyrolysis reaction was also calculated at the same level. The initial pyrolysis temperature of benzothiophene and thiophene may be close, but the pyrolysis rate of thiophene is higher than that of benzothiophene. The implication of the benzothiophene pyrolysis mechanism may be beneficial for the development of new desulfurization technology.
中文翻译:

DFT对苯并噻吩热解反应机理的研究
噻吩硫是石油焦中最稳定的有机硫,其中苯并噻吩占很大一部分。去除苯并噻吩将有助于获得超低脱硫率。在这项工作中,采用密度泛函理论(DFT)方法来研究苯并噻吩的热解机理。已发现,苯并噻吩最可能的热解反应是由α-H迁移至β位置引发的。主要产物是S自由基和乙撑硫酮,这可以很好地解释苯并噻吩的热解实验。将稠合在芳香族化合物上的噻吩转化为硫醇基可以帮助促进脱硫。相比之下,噻吩热解反应也以相同水平计算。苯并噻吩和噻吩的初始热解温度可能接近,但噻吩的热解速率高于苯并噻吩。苯并噻吩的热解机理可能对新的脱硫技术的发展有益。
更新日期:2019-01-02
中文翻译:
DFT对苯并噻吩热解反应机理的研究
噻吩硫是石油焦中最稳定的有机硫,其中苯并噻吩占很大一部分。去除苯并噻吩将有助于获得超低脱硫率。在这项工作中,采用密度泛函理论(DFT)方法来研究苯并噻吩的热解机理。已发现,苯并噻吩最可能的热解反应是由α-H迁移至β位置引发的。主要产物是S自由基和乙撑硫酮,这可以很好地解释苯并噻吩的热解实验。将稠合在芳香族化合物上的噻吩转化为硫醇基可以帮助促进脱硫。相比之下,噻吩热解反应也以相同水平计算。苯并噻吩和噻吩的初始热解温度可能接近,但噻吩的热解速率高于苯并噻吩。苯并噻吩的热解机理可能对新的脱硫技术的发展有益。
































 京公网安备 11010802027423号
京公网安备 11010802027423号