当前位置:
X-MOL 学术
›
ACS Appl. Mater. Interfaces
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Mobility Evaluation of [1]Benzothieno[3,2-b][1]benzothiophene Derivatives: Limitation and Impact on Charge Transport
ACS Applied Materials & Interfaces ( IF 8.3 ) Pub Date : 2018-12-24 00:00:00 , DOI: 10.1021/acsami.8b16158 Robert Wawrzinek , Jan Sobus , Mujeeb Ullah Chaudhry , Viqar Ahmad , Arnaud Grosjean , Jack K. Clegg , Ebinazar B. Namdas , Shih-Chun Lo
ACS Applied Materials & Interfaces ( IF 8.3 ) Pub Date : 2018-12-24 00:00:00 , DOI: 10.1021/acsami.8b16158 Robert Wawrzinek , Jan Sobus , Mujeeb Ullah Chaudhry , Viqar Ahmad , Arnaud Grosjean , Jack K. Clegg , Ebinazar B. Namdas , Shih-Chun Lo
|
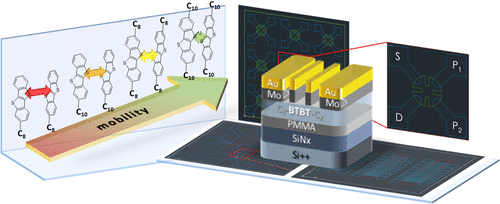
Among contemporary semiconductors, many of the best performing materials are based on [1]benzothieno[3,2-b][1]benzothiophene (BTBT). Alkylated derivatives of these small molecules not only provide high hole mobilities but also can be easily processed by thermal vacuum or solution deposition methods. Over the last decade, numerous publications have investigated molecular structures and charge transport properties to elucidate what makes these molecules so special. However, the race toward ever higher mobilities resulted in significantly deviating values, which exacerbates linking molecular structure to electronic properties. Moreover, a recently arisen debate on overestimation of organic field-effect transistor mobilities calls for a revaluation of these numbers. We synthesized and characterized four BTBT derivatives with either one or two alkyl chains (themselves consisting of either 8 or 10 carbon atoms) and investigated their spectroscopic, structural, and electrical properties. By employing two-probe, gated four-point probe and gated van der Pauw measurements, we compare field-effect mobility values at room and low temperatures and discuss their feasibility and viability. We attribute mobility changes to different angles between molecule planes and core-to-core double-layer stacking of asymmetric BTBT derivatives and show higher mobilities in the presence of more and longer alkyl chains. A so-called “zipper effect” brings BTBT cores in closer proximity promoting stronger intermolecular orbital coupling and hence higher charge transport.
中文翻译:

[1] Benzothieno [3,2- b ] [1]苯并噻吩衍生物的迁移率评估:限制及其对电荷迁移的影响
在当代半导体中,许多性能最好的材料都是基于[1] benzothieno [3,2- b] [1]苯并噻吩(BTBT)。这些小分子的烷基化衍生物不仅具有高空穴迁移率,而且可以通过热真空或溶液沉积法轻松加工。在过去的十年中,许多出版物研究了分子结构和电荷传输性质,以阐明使这些分子如此特别的原因。然而,向更高迁移率的竞争导致值显着偏离,这加剧了将分子结构与电子性质联系起来的情况。此外,最近出现的有关高估有机场效应晶体管迁移率的辩论要求重新评估这些数字。我们合成并表征了具有一个或两个烷基链(它们本身由8个或10个碳原子组成)的四种BTBT衍生物,并对其光谱进行了研究,结构和电性能。通过采用两探针,门控四点探针和门控范德堡测量,我们比较了室温和低温下的场效应迁移率值,并讨论了它们的可行性和可行性。我们将迁移率变化归因于分子平面与不对称BTBT衍生物的核对核双层堆叠之间的不同角度,并在存在更多和更长的烷基链的情况下显示出更高的迁移率。所谓的“拉链效应”使BTBT核更接近,从而促进了更强的分子间轨道耦合,从而提高了电荷传输。我们将迁移率变化归因于分子平面与不对称BTBT衍生物的核对核双层堆叠之间的不同角度,并在存在更多和更长的烷基链的情况下显示出更高的迁移率。所谓的“拉链效应”使BTBT核更接近,从而促进了更强的分子间轨道耦合,从而提高了电荷传输。我们将迁移率变化归因于分子平面与不对称BTBT衍生物的核对核双层堆叠之间的不同角度,并在存在更多和更长的烷基链的情况下显示出更高的迁移率。所谓的“拉链效应”使BTBT核更接近,从而促进了更强的分子间轨道耦合,从而提高了电荷传输。
更新日期:2018-12-24
中文翻译:
[1] Benzothieno [3,2- b ] [1]苯并噻吩衍生物的迁移率评估:限制及其对电荷迁移的影响
在当代半导体中,许多性能最好的材料都是基于[1] benzothieno [3,2- b] [1]苯并噻吩(BTBT)。这些小分子的烷基化衍生物不仅具有高空穴迁移率,而且可以通过热真空或溶液沉积法轻松加工。在过去的十年中,许多出版物研究了分子结构和电荷传输性质,以阐明使这些分子如此特别的原因。然而,向更高迁移率的竞争导致值显着偏离,这加剧了将分子结构与电子性质联系起来的情况。此外,最近出现的有关高估有机场效应晶体管迁移率的辩论要求重新评估这些数字。我们合成并表征了具有一个或两个烷基链(它们本身由8个或10个碳原子组成)的四种BTBT衍生物,并对其光谱进行了研究,结构和电性能。通过采用两探针,门控四点探针和门控范德堡测量,我们比较了室温和低温下的场效应迁移率值,并讨论了它们的可行性和可行性。我们将迁移率变化归因于分子平面与不对称BTBT衍生物的核对核双层堆叠之间的不同角度,并在存在更多和更长的烷基链的情况下显示出更高的迁移率。所谓的“拉链效应”使BTBT核更接近,从而促进了更强的分子间轨道耦合,从而提高了电荷传输。我们将迁移率变化归因于分子平面与不对称BTBT衍生物的核对核双层堆叠之间的不同角度,并在存在更多和更长的烷基链的情况下显示出更高的迁移率。所谓的“拉链效应”使BTBT核更接近,从而促进了更强的分子间轨道耦合,从而提高了电荷传输。我们将迁移率变化归因于分子平面与不对称BTBT衍生物的核对核双层堆叠之间的不同角度,并在存在更多和更长的烷基链的情况下显示出更高的迁移率。所谓的“拉链效应”使BTBT核更接近,从而促进了更强的分子间轨道耦合,从而提高了电荷传输。






























 京公网安备 11010802027423号
京公网安备 11010802027423号