当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Rigorous Free Energy Perturbation Approach to Estimating Relative Binding Affinities between Ligands with Multiple Protonation and Tautomeric States
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-12-11 00:00:00 , DOI: 10.1021/acs.jctc.8b00826 César de Oliveira 1 , Haoyu S. Yu 2 , Wei Chen 2 , Robert Abel 2 , Lingle Wang 2
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-12-11 00:00:00 , DOI: 10.1021/acs.jctc.8b00826 César de Oliveira 1 , Haoyu S. Yu 2 , Wei Chen 2 , Robert Abel 2 , Lingle Wang 2
Affiliation
|
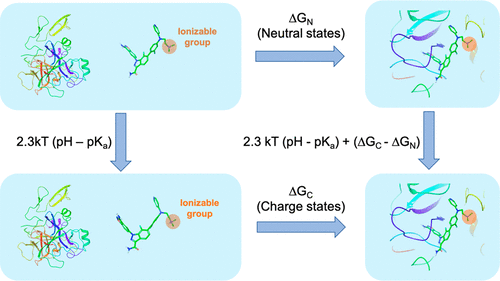
Accurate prediction of ligand binding affinities is of key importance in small molecule lead optimization and a central task in computational medicinal chemistry. Over the years, advances in both computer hardware and computational methodologies have established free energy perturbation (FEP) methods as among the most reliable and rigorous approaches to compute protein–ligand binding free energies. However, accurate description of ionization and tautomerism of ligands is still a major challenge in structure-based prediction of binding affinities. Druglike molecules are often weak acid or bases with multiple accessible protonation and tautomeric states that can contribute significantly to the binding process. To address this issue, we introduce in this work the pKa and tautomeric state correction approach. This approach is based on free energy perturbation formalism and provides a rigorous treatment of the ionization and tautomeric equilibria of ligands in solution and in the protein complexes. A series of Kinesin Spindle Protein (KSP) and Factor Xa inhibitor molecules were used as test cases. Our results demonstrate that the pKa and tautomeric state correction approach is able to rigorously and accurately incorporate multiple protonation and tautomeric states in the binding affinity calculations.
中文翻译:

严格的自由能微扰方法估计具有多个质子化和互变异构体的配体之间的相对结合亲和力
准确预测配体结合亲和力对于小分子先导物优化和计算药物化学的中心任务至关重要。多年来,计算机硬件和计算方法的进步已经建立了自由能扰动(FEP)方法,是计算蛋白质与配体结合自由能的最可靠,最严格的方法之一。然而,配体的电离和互变异构的准确描述仍然是基于结构的结合亲和力预测中的主要挑战。类药物分子通常是弱酸或具有多个可及的质子化和互变异构状态的碱,可显着促进结合过程。为了解决这个问题,我们在这项工作中介绍了p K a和互变异构状态校正方法。该方法基于自由能扰动形式,并提供溶液和蛋白质复合物中配体的电离和互变异构平衡的严格处理。一系列的驱动蛋白主轴蛋白(KSP)和Xa因子抑制剂分子被用作测试案例。我们的结果表明,p K a和互变异构状态校正方法能够在结合亲和力计算中严格而准确地合并多个质子化和互变异构状态。
更新日期:2018-12-11
中文翻译:
严格的自由能微扰方法估计具有多个质子化和互变异构体的配体之间的相对结合亲和力
准确预测配体结合亲和力对于小分子先导物优化和计算药物化学的中心任务至关重要。多年来,计算机硬件和计算方法的进步已经建立了自由能扰动(FEP)方法,是计算蛋白质与配体结合自由能的最可靠,最严格的方法之一。然而,配体的电离和互变异构的准确描述仍然是基于结构的结合亲和力预测中的主要挑战。类药物分子通常是弱酸或具有多个可及的质子化和互变异构状态的碱,可显着促进结合过程。为了解决这个问题,我们在这项工作中介绍了p K a和互变异构状态校正方法。该方法基于自由能扰动形式,并提供溶液和蛋白质复合物中配体的电离和互变异构平衡的严格处理。一系列的驱动蛋白主轴蛋白(KSP)和Xa因子抑制剂分子被用作测试案例。我们的结果表明,p K a和互变异构状态校正方法能够在结合亲和力计算中严格而准确地合并多个质子化和互变异构状态。






























 京公网安备 11010802027423号
京公网安备 11010802027423号