当前位置:
X-MOL 学术
›
Chem. Mater.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Do Chalcogenide Double Perovskites Work as Solar Cell Absorbers: A First-Principles Study
Chemistry of Materials ( IF 7.2 ) Pub Date : 2018-12-10 00:00:00 , DOI: 10.1021/acs.chemmater.8b04320
Qingde Sun 1, 2 , Hangyan Chen 3 , Wan-Jian Yin 1, 2
Chemistry of Materials ( IF 7.2 ) Pub Date : 2018-12-10 00:00:00 , DOI: 10.1021/acs.chemmater.8b04320
Qingde Sun 1, 2 , Hangyan Chen 3 , Wan-Jian Yin 1, 2
Affiliation
|
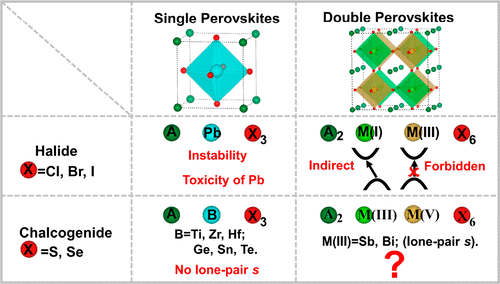
Organic–inorganic hybrid perovskite solar cells have recently been developed at an unprecedented rate as an emerging solar cell technology, with its certified power conversion efficiency (PCE) (23.7%) surpassing conventional thin-film contenders. However, the poor long-term stability and toxicity of Pb pose major setbacks to its commercialization. Theoretical calculations and experimental trail-and-error processes have recently aimed to find alternative perovskites, including inorganic halide perovskites (CsPbI3, CsPbIBr2, etc.), inorganic halide double perovskites (Cs2AgBiBr6, etc.), and chalcogenide single perovskites (BaZrS3, etc.). However, their material properties are inferior to hybrid perovskite in terms of cell performance and material toxicity. Here, a class of lead-free chalcogenide double perovskites A2M(III)M(V)X6 [A = Ca2+, Sr2+, Ba2+; M(III) = Bi3+ or Sb3+; M(V) = V5+, Nb5+, Ta5+; X = S2–, Se2–] are comprehensively investigated with respect to its stability and electronic and optical properties. First-principles calculations on bandgaps, effective masses, optical absorption, and ideal power conversion efficiencies led to the selection of nine stable double chalcogenide perovskites that exhibit superior optoelectronic properties, i.e., quasi-direct bandgaps, balanced electron and hole effective masses, and strong optical absorption owing to the strong antibonding character both at the valence band maximum (VBM) and conduction band minimum (CBM). Unfortunately, thermodynamic stability calculations on massive decomposition pathways show negative decomposition energies ranging from 0 (−0.37) to −66 meV/atom, indicating the difficulty for a thin-film phase. The most likely compound is Ba2BiNbS6, with its decomposition energies (0 and −22 meV/atom for P21/n and R3̅ phases, respectively) within the computational errors, which may be further stabilized by the confinement effect in nanocrystal form.
中文翻译:

硫属化物双钙钛矿能用作太阳能电池吸收剂吗?第一原理研究
作为一种新兴的太阳能电池技术,有机-无机混合钙钛矿太阳能电池最近得到了空前的发展,其认证的功率转换效率(PCE)(23.7%)超过了传统的薄膜竞争者。但是,Pb的长期稳定性和毒性差,使其商业化受到重大挫折。理论计算和实验误差过程最近旨在寻找替代的钙钛矿,包括无机卤化物钙钛矿(CsPbI 3,CsPbIBr 2等),无机卤化物双钙钛矿(Cs 2 AgBiBr 6等)和单硫族化物钙钛矿(BaZrS 3, ETC。)。但是,就细胞性能和材料毒性而言,它们的材料性能不如杂钙钛矿。在此,一类无铅硫族化物双钙钛矿A 2 M(III)M(V)X 6 [A = Ca 2 +,Sr 2 +,Ba 2+;M(III)= Bi 3+或Sb 3+ ; M(V)= V 5+,Nb 5+,Ta 5+。X = S 2–,Se 2–关于其稳定性以及电子和光学性质,对其进行了全面研究。通过对带隙,有效质量,光吸收和理想功率转换效率的第一性原理的计算,我们选择了九种稳定的双硫族化物钙钛矿,这些钙钛矿具有优异的光电性能,即准直接带隙,平衡的电子和空穴有效质量,并且强度高。由于在价带最大值(VBM)和导带最小值(CBM)均具有很强的抗粘结特性,因此具有良好的光吸收性能。不幸的是,大量分解路径的热力学稳定性计算显示负分解能范围为0(-0.37)至-66 meV /原子,这说明薄膜相的难度。最可能的化合物是Ba 2 BiNbS6,其分解能(0和-22兆电子伏/原子为P 2 1 / Ñ和- [R 3个阶段,分别地)的计算误差,其可通过在纳米晶体形式的限制效应进一步稳定内。
更新日期:2018-12-10
中文翻译:
硫属化物双钙钛矿能用作太阳能电池吸收剂吗?第一原理研究
作为一种新兴的太阳能电池技术,有机-无机混合钙钛矿太阳能电池最近得到了空前的发展,其认证的功率转换效率(PCE)(23.7%)超过了传统的薄膜竞争者。但是,Pb的长期稳定性和毒性差,使其商业化受到重大挫折。理论计算和实验误差过程最近旨在寻找替代的钙钛矿,包括无机卤化物钙钛矿(CsPbI 3,CsPbIBr 2等),无机卤化物双钙钛矿(Cs 2 AgBiBr 6等)和单硫族化物钙钛矿(BaZrS 3, ETC。)。但是,就细胞性能和材料毒性而言,它们的材料性能不如杂钙钛矿。在此,一类无铅硫族化物双钙钛矿A 2 M(III)M(V)X 6 [A = Ca 2 +,Sr 2 +,Ba 2+;M(III)= Bi 3+或Sb 3+ ; M(V)= V 5+,Nb 5+,Ta 5+。X = S 2–,Se 2–关于其稳定性以及电子和光学性质,对其进行了全面研究。通过对带隙,有效质量,光吸收和理想功率转换效率的第一性原理的计算,我们选择了九种稳定的双硫族化物钙钛矿,这些钙钛矿具有优异的光电性能,即准直接带隙,平衡的电子和空穴有效质量,并且强度高。由于在价带最大值(VBM)和导带最小值(CBM)均具有很强的抗粘结特性,因此具有良好的光吸收性能。不幸的是,大量分解路径的热力学稳定性计算显示负分解能范围为0(-0.37)至-66 meV /原子,这说明薄膜相的难度。最可能的化合物是Ba 2 BiNbS6,其分解能(0和-22兆电子伏/原子为P 2 1 / Ñ和- [R 3个阶段,分别地)的计算误差,其可通过在纳米晶体形式的限制效应进一步稳定内。
































 京公网安备 11010802027423号
京公网安备 11010802027423号