当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Insight into the Vibrational and Thermodynamic Properties of Layered Lithium Transition-Metal Oxides LiMO2 (M = Co, Ni, Mn): A First-Principles Study
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2016-03-10 00:00:00 , DOI: 10.1021/acs.jpcc.5b11891
Taoyuan Du 1 , Bo Xu 1 , Musheng Wu 1 , Gang Liu 1 , Chuying Ouyang 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2016-03-10 00:00:00 , DOI: 10.1021/acs.jpcc.5b11891
Taoyuan Du 1 , Bo Xu 1 , Musheng Wu 1 , Gang Liu 1 , Chuying Ouyang 1
Affiliation
|
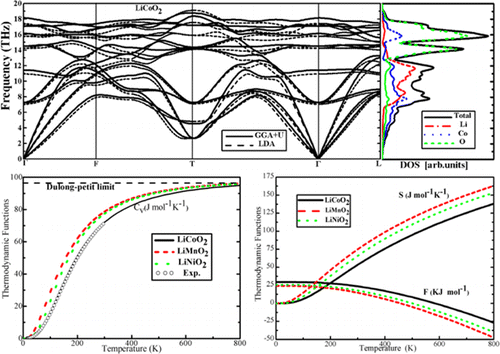
Evaluation of the finite-temperature thermodynamic properties of the electrode materials generally helps to accurately describe the performance of Li-ion battery (LIBs). To know the characteristics of the layered lithium transition-metal oxides LiMO2 (M = Co, Ni, Mn) comprehensively, herein, the vibrational and related thermodynamic quantities of these electrode materials are investigated by using density functional perturbation theory (DFPT). Local density approximation (LDA) and generalized gradient approximation with the Hubbard model correction (GGA+U) yield similar results, either for the phonon dispersion or for the thermodynamic functions. Among the three layered lithium transition-metal oxides, the vibrational and thermodynamic properties of LiNiO2 is more close to that of LiMnO2, while relatively far away from that of LiCoO2, due to the same crystal structure of LiNiO2 and LiMnO2, which is different from that of LiCoO2. In addition, the corrections of average intercalation voltage as a function of temperature for Li0.75CoO2 and Li0.5CoO2 are evaluated when considering the contribution of vibrational entropy. Since our theoretical results for LiCoO2 agree well with those from experiments, we can provide the reliable thermodynamic data for the layered lithium transition-metal oxides.
中文翻译:

层状锂过渡金属氧化物LiMO 2(M = Co,Ni,Mn)的振动和热力学性质的洞察:第一性原理研究
评估电极材料的有限温度热力学特性通常有助于准确描述锂离子电池(LIB)的性能。为了全面了解层状锂过渡金属氧化物LiMO 2(M = Co,Ni,Mn)的特性,在本文中,通过使用密度泛函扰动理论(DFPT)研究了这些电极材料的振动量和相关的热力学量。对于声子色散或热力学函数,使用Hubbard模型校正(GGA + U)的局部密度近似(LDA)和广义梯度近似产生相似的结果。在三层锂过渡金属氧化物中,LiNiO 2的振动和热力学性质更接近的LiMnO的2,而相对远离该的LiCoO 2,由于的LiNiO的相同的晶体结构2和的LiMnO 2,其是来自的LiCoO的不同2。此外,在考虑振动熵的影响时,评估了Li 0.75 CoO 2和Li 0.5 CoO 2的平均嵌入电压随温度的校正。由于我们对LiCoO 2的理论结果与实验结果吻合良好,因此我们可以为层状锂过渡金属氧化物提供可靠的热力学数据。
更新日期:2016-03-10
中文翻译:
层状锂过渡金属氧化物LiMO 2(M = Co,Ni,Mn)的振动和热力学性质的洞察:第一性原理研究
评估电极材料的有限温度热力学特性通常有助于准确描述锂离子电池(LIB)的性能。为了全面了解层状锂过渡金属氧化物LiMO 2(M = Co,Ni,Mn)的特性,在本文中,通过使用密度泛函扰动理论(DFPT)研究了这些电极材料的振动量和相关的热力学量。对于声子色散或热力学函数,使用Hubbard模型校正(GGA + U)的局部密度近似(LDA)和广义梯度近似产生相似的结果。在三层锂过渡金属氧化物中,LiNiO 2的振动和热力学性质更接近的LiMnO的2,而相对远离该的LiCoO 2,由于的LiNiO的相同的晶体结构2和的LiMnO 2,其是来自的LiCoO的不同2。此外,在考虑振动熵的影响时,评估了Li 0.75 CoO 2和Li 0.5 CoO 2的平均嵌入电压随温度的校正。由于我们对LiCoO 2的理论结果与实验结果吻合良好,因此我们可以为层状锂过渡金属氧化物提供可靠的热力学数据。
































 京公网安备 11010802027423号
京公网安备 11010802027423号