Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
An accurate TMT-based approach to quantify and model lysine susceptibility to conjugation via N-hydroxysuccinimide esters in a monoclonal antibody.
Scientific Reports ( IF 3.8 ) Pub Date : 2018-Dec-05 , DOI: 10.1038/s41598-018-35924-0 Jennifer J. Hill , Tammy-Lynn Tremblay , Christopher R. Corbeil , Enrico O. Purisima , Traian Sulea
Scientific Reports ( IF 3.8 ) Pub Date : 2018-Dec-05 , DOI: 10.1038/s41598-018-35924-0 Jennifer J. Hill , Tammy-Lynn Tremblay , Christopher R. Corbeil , Enrico O. Purisima , Traian Sulea
|
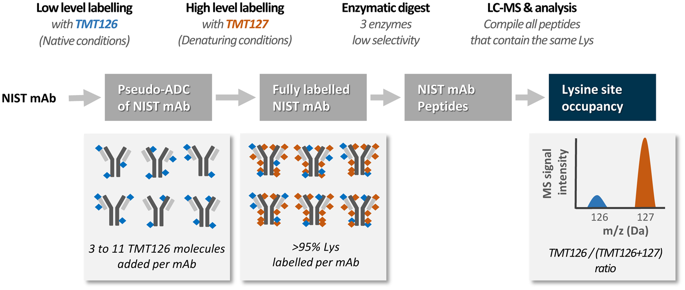
Conjugation of small molecules to proteins through N-hydroxysuccinimide (NHS) esters results in a random distribution of small molecules on lysine residues and the protein N-terminus. While mass spectrometry methods have improved characterization of these protein conjugates, it remains a challenge to quantify the occupancy at individual sites of conjugation. Here, we present a method using Tandem Mass Tags (TMT) that enabled the accurate and sensitive quantification of occupancy at individual conjugation sites in the NIST monoclonal antibody. At conjugation levels relevant to antibody drug conjugates in the clinic, site occupancy data was obtained for 37 individual sites, with average site occupancy data across 2 adjacent lysines obtained for an additional 12 sites. Thus, altogether, a measure of site occupancy was obtained for 98% of the available primary amines. We further showed that removal of the Fc-glycan on the NIST mAb increased conjugation at two specific sites in the heavy chain, demonstrating the utility of this method to identify changes in the susceptibility of individual sites to conjugation. This improved site occupancy data allowed calibration of a bi-parametric linear model for predicting the susceptibility of individual lysines to conjugation from 3D-structure based on their solvent exposures and ionization constants. Trained against the experimental data for lysines from the Fab fragment, the model provided accurate predictions of occupancies at lysine sites from the Fc region and the protein N-terminus (R2 = 0.76). This predictive model will enable improved engineering of antibodies for optimal labeling with fluorophores, toxins, or crosslinkers.
中文翻译:

一种精确的基于TMT的方法,用于定量和建模赖氨酸对单克隆抗体中通过N-羟基琥珀酰亚胺酯缀合的敏感性。
小分子通过N-羟基琥珀酰亚胺(NHS)酯与蛋白质结合,导致小分子在赖氨酸残基和N末端蛋白上随机分布。尽管质谱方法已改善了这些蛋白质结合物的表征,但量化结合处各个位点的占有率仍然是一个挑战。在这里,我们提出了一种使用串联质谱标签(TMT)的方法,该方法能够对NIST单克隆抗体中各个缀合位点的占有率进行准确而灵敏的定量。在临床上与抗体药物偶联物相关的结合水平下,获得了37个独立位点的位点占用数据,另外2个相邻赖氨酸的平均位点占用数据则另外获得了12个位点。因此,总的来说,98%的可用伯胺获得了一定的位置占用率。我们进一步显示,去除NIST mAb上的Fc-聚糖会增加重链中两个特定位点的结合,这表明该方法可用于鉴定单个位点对结合的敏感性的变化。这种改进的位点占用数据允许校准双参数线性模型,该模型用于基于其溶剂暴露量和电离常数来预测各个赖氨酸与3D结构结合的敏感性。根据来自Fab片段的赖氨酸的实验数据进行训练,该模型提供了Fc区和蛋白N端(R 我们进一步显示,去除NIST mAb上的Fc-聚糖会增加重链中两个特定位点的结合,这表明该方法可用于鉴定单个位点对结合的敏感性的变化。这种改进的位点占用数据允许校准双参数线性模型,该模型用于基于其溶剂暴露量和电离常数来预测各个赖氨酸与3D结构结合的敏感性。根据来自Fab片段的赖氨酸的实验数据进行训练,该模型提供了Fc区和蛋白N端(R 我们进一步显示,去除NIST mAb上的Fc-聚糖会增加重链中两个特定位点的结合,这表明该方法可用于鉴定单个位点对结合的敏感性的变化。这种改进的位点占用数据允许校准双参数线性模型,该模型用于基于其溶剂暴露量和电离常数来预测各个赖氨酸与3D结构结合的敏感性。根据来自Fab片段的赖氨酸的实验数据进行训练,该模型提供了Fc区和蛋白N端(R 这种改进的位点占用数据允许校准双参数线性模型,该模型用于基于其溶剂暴露量和电离常数来预测各个赖氨酸与3D结构结合的敏感性。根据来自Fab片段的赖氨酸的实验数据进行训练,该模型提供了Fc区和蛋白N端(R 这种改进的位点占用数据允许校准双参数线性模型,该模型用于基于其溶剂暴露量和电离常数来预测各个赖氨酸与3D结构结合的敏感性。根据来自Fab片段的赖氨酸的实验数据进行训练,该模型提供了Fc区和蛋白N端(R2 = 0.76)。这种预测模型将能够改进抗体的工程设计,以最佳地标记荧光团,毒素或交联剂。
更新日期:2018-12-05
中文翻译:
一种精确的基于TMT的方法,用于定量和建模赖氨酸对单克隆抗体中通过N-羟基琥珀酰亚胺酯缀合的敏感性。
小分子通过N-羟基琥珀酰亚胺(NHS)酯与蛋白质结合,导致小分子在赖氨酸残基和N末端蛋白上随机分布。尽管质谱方法已改善了这些蛋白质结合物的表征,但量化结合处各个位点的占有率仍然是一个挑战。在这里,我们提出了一种使用串联质谱标签(TMT)的方法,该方法能够对NIST单克隆抗体中各个缀合位点的占有率进行准确而灵敏的定量。在临床上与抗体药物偶联物相关的结合水平下,获得了37个独立位点的位点占用数据,另外2个相邻赖氨酸的平均位点占用数据则另外获得了12个位点。因此,总的来说,98%的可用伯胺获得了一定的位置占用率。我们进一步显示,去除NIST mAb上的Fc-聚糖会增加重链中两个特定位点的结合,这表明该方法可用于鉴定单个位点对结合的敏感性的变化。这种改进的位点占用数据允许校准双参数线性模型,该模型用于基于其溶剂暴露量和电离常数来预测各个赖氨酸与3D结构结合的敏感性。根据来自Fab片段的赖氨酸的实验数据进行训练,该模型提供了Fc区和蛋白N端(R 我们进一步显示,去除NIST mAb上的Fc-聚糖会增加重链中两个特定位点的结合,这表明该方法可用于鉴定单个位点对结合的敏感性的变化。这种改进的位点占用数据允许校准双参数线性模型,该模型用于基于其溶剂暴露量和电离常数来预测各个赖氨酸与3D结构结合的敏感性。根据来自Fab片段的赖氨酸的实验数据进行训练,该模型提供了Fc区和蛋白N端(R 我们进一步显示,去除NIST mAb上的Fc-聚糖会增加重链中两个特定位点的结合,这表明该方法可用于鉴定单个位点对结合的敏感性的变化。这种改进的位点占用数据允许校准双参数线性模型,该模型用于基于其溶剂暴露量和电离常数来预测各个赖氨酸与3D结构结合的敏感性。根据来自Fab片段的赖氨酸的实验数据进行训练,该模型提供了Fc区和蛋白N端(R 这种改进的位点占用数据允许校准双参数线性模型,该模型用于基于其溶剂暴露量和电离常数来预测各个赖氨酸与3D结构结合的敏感性。根据来自Fab片段的赖氨酸的实验数据进行训练,该模型提供了Fc区和蛋白N端(R 这种改进的位点占用数据允许校准双参数线性模型,该模型用于基于其溶剂暴露量和电离常数来预测各个赖氨酸与3D结构结合的敏感性。根据来自Fab片段的赖氨酸的实验数据进行训练,该模型提供了Fc区和蛋白N端(R2 = 0.76)。这种预测模型将能够改进抗体的工程设计,以最佳地标记荧光团,毒素或交联剂。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号