当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Theoretical Approaches to Describing the Oxygen Reduction Reaction Activity of Single-Atom Catalysts
The Journal of Physical Chemistry C ( IF 3.2 ) Pub Date : 2018-12-18 , DOI: 10.1021/acs.jpcc.8b09430
Anjli M. Patel , Stefan Ringe , Samira Siahrostami , Michal Bajdich , Jens K. Nørskov , Ambarish R. Kulkarni
The Journal of Physical Chemistry C ( IF 3.2 ) Pub Date : 2018-12-18 , DOI: 10.1021/acs.jpcc.8b09430
Anjli M. Patel , Stefan Ringe , Samira Siahrostami , Michal Bajdich , Jens K. Nørskov , Ambarish R. Kulkarni
|
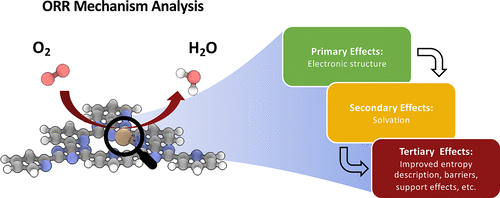
Single-atom catalysts have recently emerged as promising low-cost alternatives to Pt for the oxygen reduction reaction (ORR). Given the unique properties that distinguish these systems from traditional transition-metal electrocatalysts, it is essential to benchmark and establish appropriate computational approaches to study these novel materials. Herein, we employ multiple levels of theory, including wave function methods, density functional theory (DFT), and classical simulations, to investigate Cu-modified covalent triazine framework catalysts (Cu/CTF). We consider three major aspects of treating this system computationally. First, we present a step-wise approach to predict the ORR mechanism and adsorbate coverages on Cu/CTF. We then benchmark various DFT methods to coupled-cluster theory with the domain-based local pair natural orbital approximation, which indicates that HSE06 and PBE0 hybrid functionals most accurately describe the adsorption energies of ORR adsorbates on Cu/CTF. We finally employ thermodynamic integration and other techniques to consider solvation effects, which play significant roles in predicting the energies of reaction intermediates and the overall ORR pathway. Our findings indicate that accurate descriptions of both the electronic structure and solvation are necessary to understand the ORR activity of Cu/CTF.
中文翻译:

描述单原子催化剂氧还原反应活性的理论方法
最近,单原子催化剂作为氧还原反应(ORR)的有前景的低成本替代品出现了。鉴于将这些系统与传统的过渡金属电催化剂区分开来的独特特性,必须进行基准测试并建立适当的计算方法来研究这些新型材料,这一点至关重要。在本文中,我们采用了多种层次的理论,包括波函数方法,密度泛函理论(DFT)和经典模拟,来研究Cu改性的共价三嗪骨架催化剂(Cu / CTF)。我们考虑在计算上处理该系统的三个主要方面。首先,我们提出一种逐步方法来预测ORR机理和Cu / CTF的吸附物覆盖率。然后,我们使用基于域的局部对自然轨道逼近,将各种DFT方法与耦合簇理论进行了比较,这表明HSE06和PBE0杂合功能最准确地描述了ORR在Cu / CTF上的吸附能。我们最终采用热力学积分和其他技术来考虑溶剂化作用,这些作用在预测反应中间体的能量和整个ORR途径中起着重要作用。我们的发现表明,对电子结构和溶剂化的准确描述对于理解Cu / CTF的ORR活性是必要的。在预测反应中间体的能量和整个ORR途径中起重要作用。我们的发现表明,对电子结构和溶剂化的准确描述对于理解Cu / CTF的ORR活性是必要的。在预测反应中间体的能量和整个ORR途径中起重要作用。我们的发现表明,对电子结构和溶剂化的准确描述对于理解Cu / CTF的ORR活性是必要的。
更新日期:2018-12-19
中文翻译:
描述单原子催化剂氧还原反应活性的理论方法
最近,单原子催化剂作为氧还原反应(ORR)的有前景的低成本替代品出现了。鉴于将这些系统与传统的过渡金属电催化剂区分开来的独特特性,必须进行基准测试并建立适当的计算方法来研究这些新型材料,这一点至关重要。在本文中,我们采用了多种层次的理论,包括波函数方法,密度泛函理论(DFT)和经典模拟,来研究Cu改性的共价三嗪骨架催化剂(Cu / CTF)。我们考虑在计算上处理该系统的三个主要方面。首先,我们提出一种逐步方法来预测ORR机理和Cu / CTF的吸附物覆盖率。然后,我们使用基于域的局部对自然轨道逼近,将各种DFT方法与耦合簇理论进行了比较,这表明HSE06和PBE0杂合功能最准确地描述了ORR在Cu / CTF上的吸附能。我们最终采用热力学积分和其他技术来考虑溶剂化作用,这些作用在预测反应中间体的能量和整个ORR途径中起着重要作用。我们的发现表明,对电子结构和溶剂化的准确描述对于理解Cu / CTF的ORR活性是必要的。在预测反应中间体的能量和整个ORR途径中起重要作用。我们的发现表明,对电子结构和溶剂化的准确描述对于理解Cu / CTF的ORR活性是必要的。在预测反应中间体的能量和整个ORR途径中起重要作用。我们的发现表明,对电子结构和溶剂化的准确描述对于理解Cu / CTF的ORR活性是必要的。



















































 京公网安备 11010802027423号
京公网安备 11010802027423号