当前位置:
X-MOL 学术
›
Cell Death Differ.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Dclk1 in tuft cells promotes inflammation-driven epithelial restitution and mitigates chronic colitis.
Cell Death and Differentiation ( IF 13.7 ) Pub Date : 2018-11-26 , DOI: 10.1038/s41418-018-0237-x
Jun Yi 1, 2 , Kirk Bergstrom 2 , Jianxin Fu 2 , Xindi Shan 2 , J Michael McDaniel 2 , Samuel McGee 2 , Dongfeng Qu 3 , Courtney W Houchen 3 , Xiaowei Liu 1 , Lijun Xia 2, 4
Cell Death and Differentiation ( IF 13.7 ) Pub Date : 2018-11-26 , DOI: 10.1038/s41418-018-0237-x
Jun Yi 1, 2 , Kirk Bergstrom 2 , Jianxin Fu 2 , Xindi Shan 2 , J Michael McDaniel 2 , Samuel McGee 2 , Dongfeng Qu 3 , Courtney W Houchen 3 , Xiaowei Liu 1 , Lijun Xia 2, 4
Affiliation
|
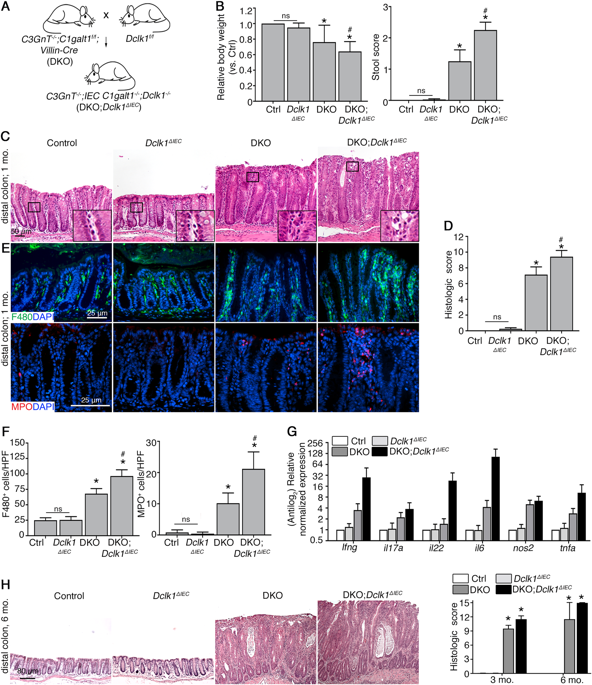
Ulcerative colitis (UC) is a chronic inflammatory bowel disease characterized by defective intestinal barrier integrity toward the microbiota and epithelial damage. Double cortin-like kinase 1 (Dclk1), a marker of intestinal tuft cells, can regulate tissue regenerative responses, but its role in epithelial repair during bacterial-dependent chronic colitis is unclear. We addressed this question using our recently developed mouse model of spontaneous microbiota-dependent colitis induced by mucin-type O-glycan deficiency (DKO), which recapitulates most features of human UC. We generated DKO mice lacking intestinal epithelial Dclk1 (DKO;Dclk1ΔIEC) and analyzed colitis onset and severity using clinical and histologic indices, immune responses by qPCR and immunostaining, and epithelial responses using proliferation markers and organoid culture. We found 3-4-week-old DKO;Dclk1ΔIEC mice developed worsened spontaneous colitis characterized by reduced body weight, loose stool, severe colon thickening, epithelial lesions, and inflammatory cell infiltrates compared with DKO mice. The primary defect was an impaired epithelial proliferative response during inflammation. Dclk1 deficiency also reduced inflammation-induced proliferation and growth of colon organoids ex vivo. Mechanistically, Dclk1 expression was important for inflammation-induced Cox2 expression and prostaglandin E2 (PGE2) production in vivo, and PGE2 rescued proliferative defects in Dclk1-deficient colonic organoids. Although tuft cells were expanded in both DKO and DKO;Dclk1ΔIEC relative to WT mice, loss of Dclk1 was associated with reduced tuft cell activation (i.e., proliferation) during inflammation. Similar results were found in DKO vs. DKO;Dclk1ΔIEC mice at 3-6 months of age. Our results support that tuft cells, via Dclk1, are important responders to bacterial-induced colitis by enhancing epithelial repair responses, which in turn limits bacterial infiltration into the mucosa.
中文翻译:

簇细胞中的 Dclk1 促进炎症驱动的上皮恢复并减轻慢性结肠炎。
溃疡性结肠炎(UC)是一种慢性炎症性肠病,其特征是肠道微生物群屏障完整性缺陷和上皮损伤。双皮质蛋白样激酶 1 (Dclk1) 是肠簇细胞的标记物,可以调节组织再生反应,但其在细菌依赖性慢性结肠炎期间上皮修复中的作用尚不清楚。我们使用最近开发的由粘蛋白型 O-聚糖缺乏 (DKO) 诱导的自发性微生物依赖型结肠炎小鼠模型来解决这个问题,该模型概括了人类 UC 的大多数特征。我们生成了缺乏肠上皮 Dclk1 (DKO;Dclk1ΔIEC) 的 DKO 小鼠,并使用临床和组织学指标分析结肠炎的发病和严重程度,通过 qPCR 和免疫染色分析免疫反应,并使用增殖标记物和类器官培养分析上皮反应。我们发现,与 DKO 小鼠相比,3-4 周龄的 DKO;Dclk1ΔIEC 小鼠出现了更严重的自发性结肠炎,其特征是体重减轻、稀便、结肠严重增厚、上皮病变和炎症细胞浸润。主要缺陷是炎症期间上皮增殖反应受损。 Dclk1 缺乏还减少了炎症诱导的离体结肠类器官的增殖和生长。从机制上讲,Dclk1 表达对于体内炎症诱导的 Cox2 表达和前列腺素 E2 (PGE2) 产生很重要,PGE2 可以挽救 Dclk1 缺陷的结肠类器官的增殖缺陷。尽管相对于 WT 小鼠,DKO 和 DKO;Dclk1ΔIEC 中的簇细胞均扩增,但 Dclk1 的缺失与炎症期间簇细胞活化(即增殖)的减少相关。在 3-6 个月龄的 DKO 与 DKO;Dclk1ΔIEC 小鼠中也发现了类似的结果。 我们的结果支持簇细胞通过 Dclk1 增强上皮修复反应,从而限制细菌渗入粘膜,从而成为细菌诱导的结肠炎的重要应答者。
更新日期:2019-01-26
中文翻译:
簇细胞中的 Dclk1 促进炎症驱动的上皮恢复并减轻慢性结肠炎。
溃疡性结肠炎(UC)是一种慢性炎症性肠病,其特征是肠道微生物群屏障完整性缺陷和上皮损伤。双皮质蛋白样激酶 1 (Dclk1) 是肠簇细胞的标记物,可以调节组织再生反应,但其在细菌依赖性慢性结肠炎期间上皮修复中的作用尚不清楚。我们使用最近开发的由粘蛋白型 O-聚糖缺乏 (DKO) 诱导的自发性微生物依赖型结肠炎小鼠模型来解决这个问题,该模型概括了人类 UC 的大多数特征。我们生成了缺乏肠上皮 Dclk1 (DKO;Dclk1ΔIEC) 的 DKO 小鼠,并使用临床和组织学指标分析结肠炎的发病和严重程度,通过 qPCR 和免疫染色分析免疫反应,并使用增殖标记物和类器官培养分析上皮反应。我们发现,与 DKO 小鼠相比,3-4 周龄的 DKO;Dclk1ΔIEC 小鼠出现了更严重的自发性结肠炎,其特征是体重减轻、稀便、结肠严重增厚、上皮病变和炎症细胞浸润。主要缺陷是炎症期间上皮增殖反应受损。 Dclk1 缺乏还减少了炎症诱导的离体结肠类器官的增殖和生长。从机制上讲,Dclk1 表达对于体内炎症诱导的 Cox2 表达和前列腺素 E2 (PGE2) 产生很重要,PGE2 可以挽救 Dclk1 缺陷的结肠类器官的增殖缺陷。尽管相对于 WT 小鼠,DKO 和 DKO;Dclk1ΔIEC 中的簇细胞均扩增,但 Dclk1 的缺失与炎症期间簇细胞活化(即增殖)的减少相关。在 3-6 个月龄的 DKO 与 DKO;Dclk1ΔIEC 小鼠中也发现了类似的结果。 我们的结果支持簇细胞通过 Dclk1 增强上皮修复反应,从而限制细菌渗入粘膜,从而成为细菌诱导的结肠炎的重要应答者。
































 京公网安备 11010802027423号
京公网安备 11010802027423号