当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Relative rates of hydrogen shift isomerizations depend strongly on multiple-structure anharmonicity
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2018-11-23 , DOI: 10.1021/jacs.8b09381 Lili Xing 1 , Junwei Lucas Bao 2 , Zhandong Wang 3 , Xuetao Wang 1 , Donald G. Truhlar 2
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2018-11-23 , DOI: 10.1021/jacs.8b09381 Lili Xing 1 , Junwei Lucas Bao 2 , Zhandong Wang 3 , Xuetao Wang 1 , Donald G. Truhlar 2
Affiliation
|
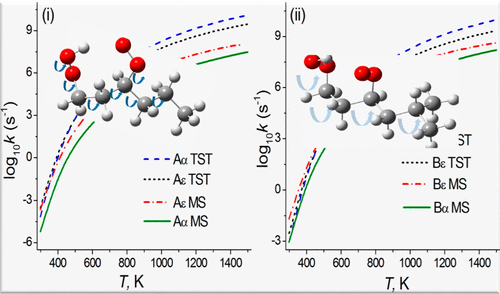
Hydroperoxyalkylperoxy species (OOQOOH) are important intermediates that are generated during the autoignition of transport fuels. A key reaction of hydroperoxyalkylperoxy radicals is a [1,5] hydrogen shift, for which kinetics data are experimentally unavailable. Here we study two typical OOQOOH reactions and compare their kinetics to one another and to a previous study to learn the effect of structural variations of the alkyl group on the competition between alternative [1,5] hydrogen shifts of hydroperoxyalkylperoxy species. We use electronic structure calculations to determine previously missing thermochemical data, and we use variational transition state theory with multidimensional tunneling, multiple structures, torsional potential anharmonicity, and high-frequency anharmonicity to obtain more accurate rate constants than the ones that can be computed by conventional single-structure harmonic transition state theory and than the empirically estimated rate constants that are currently used in combustion modeling. The calculated temperature range is 298-1500 K. The roles of various factors in determining the rates are elucidated, and we find an especially strong effect of multiple structure anharmonicity due to torsions. Thus, even though there is some cancellation between the anharmonicity of the reactant and the anharmonicity of the transition state, and even though the reactants are very similar in structure, differing only by a methyl group, the effect of multiple structure anharmonicity has a large effect on the relative rates, as large as a factor of 17 at room temperature and as large as a factor of 7 at 1500 K. This has broad implications for the estimation of reaction rates in many subfields of chemistry, including combustion chemistry and atmospheric chemistry, where rates of reaction of complex molecules are usually estimated without explicit consideration of this fundamental entropic effect. In addition, the pressure-dependence of the rate constants is modeled by system-specific quantum Rice-Ramsperger-Kassel theory for a reversible isomerization.
中文翻译:

氢位移异构化的相对速率强烈依赖于多结构非谐性
氢过氧烷基过氧物质 (OOQOOH) 是运输燃料自燃过程中产生的重要中间体。氢过氧烷基过氧自由基的一个关键反应是 [1,5] 氢位移,其动力学数据在实验上无法获得。在这里,我们研究了两个典型的 OOQOOH 反应,并将它们的动力学相互比较,并与之前的研究进行比较,以了解烷基的结构变化对氢过氧烷基过氧物种的替代 [1,5] 氢位移之间竞争的影响。我们使用电子结构计算来确定先前缺失的热化学数据,我们使用变分过渡态理论与多维隧道效应、多重结构、扭势非谐性、和高频非谐性以获得比传统单结构谐波过渡态理论可以计算的速率常数和目前用于燃烧建模的经验估计速率常数更准确的速率常数。计算出的温度范围为 298-1500 K。阐明了各种因素在决定速率中的作用,我们发现由于扭转引起的多重结构非谐性的影响特别大。因此,即使反应物的非调和性与过渡态的非调和性之间存在某种抵消,即使反应物在结构上非常相似,仅相差一个甲基,但多重结构非调和性的影响却很大。在相对利率上,在室温下高达 17 倍,在 1500 K 时高达 7 倍。这对许多化学子领域的反应速率估计具有广泛的意义,包括燃烧化学和大气化学,其中反应速率为通常在没有明确考虑这种基本熵效应的情况下估计复杂分子。此外,速率常数的压力依赖性通过系统特定的量子 Rice-Ramsperger-Kassel 理论建模,以实现可逆异构化。
更新日期:2018-11-23
中文翻译:
氢位移异构化的相对速率强烈依赖于多结构非谐性
氢过氧烷基过氧物质 (OOQOOH) 是运输燃料自燃过程中产生的重要中间体。氢过氧烷基过氧自由基的一个关键反应是 [1,5] 氢位移,其动力学数据在实验上无法获得。在这里,我们研究了两个典型的 OOQOOH 反应,并将它们的动力学相互比较,并与之前的研究进行比较,以了解烷基的结构变化对氢过氧烷基过氧物种的替代 [1,5] 氢位移之间竞争的影响。我们使用电子结构计算来确定先前缺失的热化学数据,我们使用变分过渡态理论与多维隧道效应、多重结构、扭势非谐性、和高频非谐性以获得比传统单结构谐波过渡态理论可以计算的速率常数和目前用于燃烧建模的经验估计速率常数更准确的速率常数。计算出的温度范围为 298-1500 K。阐明了各种因素在决定速率中的作用,我们发现由于扭转引起的多重结构非谐性的影响特别大。因此,即使反应物的非调和性与过渡态的非调和性之间存在某种抵消,即使反应物在结构上非常相似,仅相差一个甲基,但多重结构非调和性的影响却很大。在相对利率上,在室温下高达 17 倍,在 1500 K 时高达 7 倍。这对许多化学子领域的反应速率估计具有广泛的意义,包括燃烧化学和大气化学,其中反应速率为通常在没有明确考虑这种基本熵效应的情况下估计复杂分子。此外,速率常数的压力依赖性通过系统特定的量子 Rice-Ramsperger-Kassel 理论建模,以实现可逆异构化。






























 京公网安备 11010802027423号
京公网安备 11010802027423号