Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
First-Principles Structural, Mechanical, and Thermodynamic Calculations of the Negative Thermal Expansion Compound Zr2(WO4)(PO4)2
ACS Omega ( IF 3.7 ) Pub Date : 2018-11-20 00:00:00 , DOI: 10.1021/acsomega.8b02456 Philippe F. Weck 1 , Eunja Kim 2 , Margaret E. Gordon 1 , Jeffery A. Greathouse 1 , Rémi Dingreville 1 , Charles R. Bryan 1
ACS Omega ( IF 3.7 ) Pub Date : 2018-11-20 00:00:00 , DOI: 10.1021/acsomega.8b02456 Philippe F. Weck 1 , Eunja Kim 2 , Margaret E. Gordon 1 , Jeffery A. Greathouse 1 , Rémi Dingreville 1 , Charles R. Bryan 1
Affiliation
|
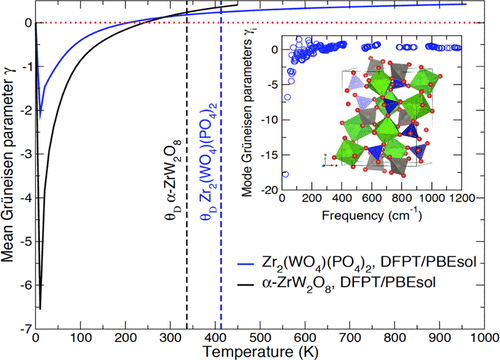
The negative thermal expansion (NTE) material Zr2(WO4)(PO4)2 has been investigated for the first time within the framework of the density functional perturbation theory (DFPT). The structural, mechanical, and thermodynamic properties of this material have been predicted using the Perdew, Burke and Ernzerhof for solid (PBEsol) exchange–correlation functional, which showed superior accuracy over standard functionals in previous computational studies of the NTE material α-ZrW2O8. The bulk modulus calculated for Zr2(WO4)(PO4)2 using the Vinet equation of state at room temperature is K0 = 63.6 GPa, which is in close agreement with the experimental estimate of 61.3(8) at T = 296 K. The computed mean linear coefficient of thermal expansion is −3.1 × 10–6 K−1 in the temperature range ∼0–70 K, in line with the X-ray diffraction measurements. The mean Grüneisen parameter controlling the thermal expansion of Zr2(WO4)(PO4)2 is negative below 205 K, with a minimum of −2.1 at 10 K. The calculated standard molar heat capacity and entropy are CP0 = 287.6 and S0 = 321.9 J·mol–1·K–1, respectively. The results reported in this study demonstrate the accuracy of DFPT/PBEsol for assessing or predicting the relationship between structural and thermomechanical properties of NTE materials.
中文翻译:

负热膨胀化合物Zr 2(WO 4)(PO 4)2的第一性原理,结构和热力学计算
负热膨胀(NTE)材料Zr 2(WO 4)(PO 4)2首次在密度泛函扰动理论(DFPT)的框架内进行了研究。这种材料的结构,机械和热力学性质已经使用Perdew,伯克和Ernzerhof为固体(PBEsol)交换相关的功能,这表明优异的准确度在NTE材料α-ZRW的先前计算的研究标准函预测2 O 8。使用室温下的维涅特状态方程计算的Zr 2(WO 4)(PO 4)2的体积模量为K 0 = 63.6 GPa,与T = 296 K时的61.3(8)实验估计值非常一致。在〜0的温度范围内,计算出的平均热膨胀线性系数为-3.1×10 –6 K -1 –70 K,符合X射线衍射测量。在205 K以下,控制Zr 2(WO 4)(PO 4)2的热膨胀的平均Grüneisen参数为负,在10 K时最小值为-2.1。计算的标准摩尔热容和熵为C P 0 = 287.6并且S 0 = 321.9 J·mol –1 ·K –1, 分别。这项研究报告的结果证明了DFPT / PBEsol用于评估或预测NTE材料的结构和热机械性能之间关系的准确性。
更新日期:2018-11-20
中文翻译:
负热膨胀化合物Zr 2(WO 4)(PO 4)2的第一性原理,结构和热力学计算
负热膨胀(NTE)材料Zr 2(WO 4)(PO 4)2首次在密度泛函扰动理论(DFPT)的框架内进行了研究。这种材料的结构,机械和热力学性质已经使用Perdew,伯克和Ernzerhof为固体(PBEsol)交换相关的功能,这表明优异的准确度在NTE材料α-ZRW的先前计算的研究标准函预测2 O 8。使用室温下的维涅特状态方程计算的Zr 2(WO 4)(PO 4)2的体积模量为K 0 = 63.6 GPa,与T = 296 K时的61.3(8)实验估计值非常一致。在〜0的温度范围内,计算出的平均热膨胀线性系数为-3.1×10 –6 K -1 –70 K,符合X射线衍射测量。在205 K以下,控制Zr 2(WO 4)(PO 4)2的热膨胀的平均Grüneisen参数为负,在10 K时最小值为-2.1。计算的标准摩尔热容和熵为C P 0 = 287.6并且S 0 = 321.9 J·mol –1 ·K –1, 分别。这项研究报告的结果证明了DFPT / PBEsol用于评估或预测NTE材料的结构和热机械性能之间关系的准确性。






























 京公网安备 11010802027423号
京公网安备 11010802027423号