当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Charge Transport in [Li(tetraglyme)][bis(trifluoromethane) sulfonimide] Solvate Ionic Liquids: Insight from Molecular Dynamics Simulations
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2018-10-18 , DOI: 10.1021/acs.jpcb.8b06913 Dengpan Dong 1 , Dmitry Bedrov 1
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2018-10-18 , DOI: 10.1021/acs.jpcb.8b06913 Dengpan Dong 1 , Dmitry Bedrov 1
Affiliation
|
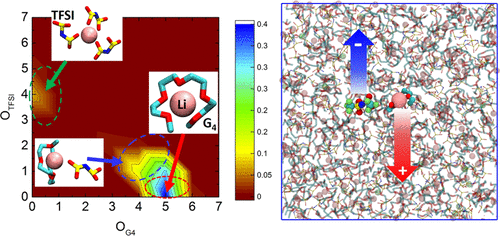
Molecular dynamics simulations using fully atomistic polarizable force field have been performed on solvate ionic liquids (SILs), comprised of tetraglyme (G4) solvent molecules, Li+ cations, and bis(trifluoromethane) sulfonimide (TFSI) anions, [Li(G4)][TFSI]. The SILs with equimolar salt:G4 composition were investigated in the 303–373 K temperature range, whereas several systems with lower salt concentrations were investigated at 373 K. The simulations using polarizable force field demonstrate very good consistency of structural and dynamic properties with experimental data. The ability to accurately sample the ion transport mechanisms is particularly encouraging, taking into account that previous simulations employing nonpolarizable models had challenges in sampling dynamics in these systems. Here, we correlate Li+ ion local environment and glyme conformations with dynamic characteristics, such as residence time of species around Li+, self-diffusion coefficients, transference number, and conductivity. The analysis of contributions to Li+ mobility due to changing its local environment (i.e., moving from one glyme/anion to another) and from translational motion of Li+ with its’ coordination environment showed significant dominance of the latter. The contributions of cross-ion dynamic correlations to the total conductivity have been quantified, showing strongly positive contribution from the cation–anion anticorrelation. Despite the high degree of Li–TFSI dissociation and positive contribution of the cation–anion anticorrelated motion to conductivity, the Li+ transference numbers for equimolar SILs are very low under the anion blocking conditions.
中文翻译:

[Li(四甘醇二甲醚)] [双(三氟甲烷)磺酰亚胺]溶剂化离子液体中的电荷传输:分子动力学模拟的见解。
已对由四甘醇二甲醚(G4)溶剂分子Li +组成的溶剂化物离子液体(SILs)进行了使用完全原子化的极化力场的分子动力学模拟阳离子和双(三氟甲烷)磺酰亚胺(TFSI)阴离子[Li(G4)] [TFSI]。在303–373 K的温度范围内研究了等摩尔盐:G4组成的SIL,而在373 K下研究了几种盐浓度较低的系统。使用极化力场进行的模拟表明,结构和动力学性质与实验数据具有很好的一致性。 。考虑到先前采用不可极化模型的模拟在这些系统中的采样动力学方面存在挑战,因此对离子迁移机制进行精确采样的能力尤其令人鼓舞。在这里,我们将Li +离子的局部环境和甘氨酸构象与动态特性(例如,Li +周围物种的停留时间)相关联,自扩散系数,转移数和电导率。由于Li +迁移环境的改变(即从一种糖蛋白/阴离子迁移到另一种)和Li +的平移运动及其配位环境而对Li +迁移率的贡献分析显示,后者具有显着优势。跨离子动态相关性对总电导率的贡献已被量化,显示出阳离子-阴离子反相关性的强正贡献。尽管Li-TFSI的解离程度很高,阳离子-阴离子反相关运动对电导率有积极贡献,但等离子SIL的Li +迁移数在阴离子阻断条件下仍然非常低。
更新日期:2018-10-19
中文翻译:
[Li(四甘醇二甲醚)] [双(三氟甲烷)磺酰亚胺]溶剂化离子液体中的电荷传输:分子动力学模拟的见解。
已对由四甘醇二甲醚(G4)溶剂分子Li +组成的溶剂化物离子液体(SILs)进行了使用完全原子化的极化力场的分子动力学模拟阳离子和双(三氟甲烷)磺酰亚胺(TFSI)阴离子[Li(G4)] [TFSI]。在303–373 K的温度范围内研究了等摩尔盐:G4组成的SIL,而在373 K下研究了几种盐浓度较低的系统。使用极化力场进行的模拟表明,结构和动力学性质与实验数据具有很好的一致性。 。考虑到先前采用不可极化模型的模拟在这些系统中的采样动力学方面存在挑战,因此对离子迁移机制进行精确采样的能力尤其令人鼓舞。在这里,我们将Li +离子的局部环境和甘氨酸构象与动态特性(例如,Li +周围物种的停留时间)相关联,自扩散系数,转移数和电导率。由于Li +迁移环境的改变(即从一种糖蛋白/阴离子迁移到另一种)和Li +的平移运动及其配位环境而对Li +迁移率的贡献分析显示,后者具有显着优势。跨离子动态相关性对总电导率的贡献已被量化,显示出阳离子-阴离子反相关性的强正贡献。尽管Li-TFSI的解离程度很高,阳离子-阴离子反相关运动对电导率有积极贡献,但等离子SIL的Li +迁移数在阴离子阻断条件下仍然非常低。




















































 京公网安备 11010802027423号
京公网安备 11010802027423号