当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Probing ATP/ATP-Aptamer or ATP-Aptamer Mutant Complexes by Microscale Thermophoresis and Molecular Dynamics Simulations: Discovery of an ATP-Aptamer Sequence of Superior Binding Properties
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2018-09-20 , DOI: 10.1021/acs.jpcb.8b06802
Yonatan Biniuri 1 , Bauke Albada 2 , Itamar Willner 1
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2018-09-20 , DOI: 10.1021/acs.jpcb.8b06802
Yonatan Biniuri 1 , Bauke Albada 2 , Itamar Willner 1
Affiliation
|
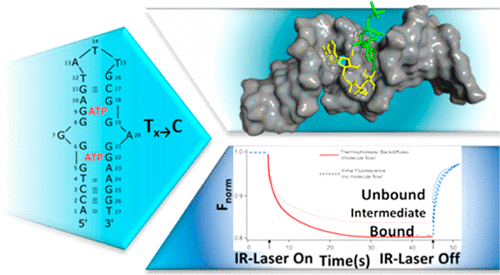
Microscale thermophoresis (MST) is used to follow the dissociation constants corresponding to ATTO 488-labeled adenosine triphosphate (ATP) and the ATP-aptamer or ATP-aptamer mutants that include two binding sites for the ATP ligand. A set of eight ATP-aptamer mutants, where the thymidine bases, within the reported ATP binding aptamer sites, are substituted with cytosine bases, are examined. The MST-derived dissociation constant of ATP to the reported aptamer is Kd = 31 ± 3 μM, whereas most of the aptamer mutants show lower affinity (higher Kd values) toward the ATP ligand. One aptamer mutant reveals, however, a higher affinity toward the ATP ligand, as compared to the reported ATP-aptamer. Molecular dynamics and docking simulations identify the structural features that control the affinities of binding of the ATP ligand to the two binding sites associated with the ATP-aptamer or the ATP-aptamer mutants. The simulated structures suggest that H-bonds between the ATP ligand and G9 and G11 bases, within one binding domain, and the π–π interactions between G6 and the ATP purine moiety and the pyrimidine ring, in the second binding domain, control the affinity of binding interactions between the ATP ligand and the ATP-aptamer or ATP-aptamer mutant. Very good correlation between the computed Kd values and the MST-derived Kd values is found. The ATP-aptamer mutant (consisting of A1→ G, T4 → C, T12 → C, A24 → G, and T27 → C mutations) reveals superior binding affinities toward the ATP ligands (Kd = 15 ± 1 μM) as compared to the binding affinity of ATP to the reported aptamer. These features of the mutant are supported by molecular dynamics simulations.
中文翻译:

探测ATP / ATP-适体或ATP-适体突变体的复合物,通过微观热泳和分子动力学模拟:ATP-适体序列具有优越的结合特性的发现
微型热泳(MST)用于追踪对应于ATTO 488标记的三磷酸腺苷(ATP)和包括两个ATP配体结合位点的ATP-适体或ATP-适体突变体的解离常数。检查了一组八个ATP适体突变体,其中已报告的ATP结合适体位点内的胸苷碱基被胞嘧啶碱基取代。MST衍生的ATP与已报道的适体的解离常数为K d = 31±3μM,而大多数适体突变体显示出较低的亲和力(较高的K d值)朝向ATP配体。然而,与报道的ATP-适体相比,一种适体突变体显示出对ATP配体的更高的亲和力。分子动力学和对接模拟确定了控制ATP配体与与ATP-适体或ATP-适体突变体相关的两个结合位点的结合亲和力的结构特征。模拟结构表明,在一个结合域中,ATP配体与G 9和G 11碱基之间存在H键,在第二个结合域中,G 6与ATP嘌呤部分和嘧啶环之间存在π–π相互作用,控制ATP配体与ATP-适体或ATP-适体突变体之间结合相互作用的亲和力。计算值之间的很好的相关性 找到K d值和MST派生的K d值。ATP适体突变体(由A 1 →G,T 4 →C,T 12 →C,A 24 →G和T 27 →C突变组成)显示出对ATP配体的优越结合亲和力(K d = 15±1 μM)与ATP与已报道的适体的结合亲和力相比。分子动力学模拟支持了突变体的这些特征。
更新日期:2018-09-21
中文翻译:
探测ATP / ATP-适体或ATP-适体突变体的复合物,通过微观热泳和分子动力学模拟:ATP-适体序列具有优越的结合特性的发现
微型热泳(MST)用于追踪对应于ATTO 488标记的三磷酸腺苷(ATP)和包括两个ATP配体结合位点的ATP-适体或ATP-适体突变体的解离常数。检查了一组八个ATP适体突变体,其中已报告的ATP结合适体位点内的胸苷碱基被胞嘧啶碱基取代。MST衍生的ATP与已报道的适体的解离常数为K d = 31±3μM,而大多数适体突变体显示出较低的亲和力(较高的K d值)朝向ATP配体。然而,与报道的ATP-适体相比,一种适体突变体显示出对ATP配体的更高的亲和力。分子动力学和对接模拟确定了控制ATP配体与与ATP-适体或ATP-适体突变体相关的两个结合位点的结合亲和力的结构特征。模拟结构表明,在一个结合域中,ATP配体与G 9和G 11碱基之间存在H键,在第二个结合域中,G 6与ATP嘌呤部分和嘧啶环之间存在π–π相互作用,控制ATP配体与ATP-适体或ATP-适体突变体之间结合相互作用的亲和力。计算值之间的很好的相关性 找到K d值和MST派生的K d值。ATP适体突变体(由A 1 →G,T 4 →C,T 12 →C,A 24 →G和T 27 →C突变组成)显示出对ATP配体的优越结合亲和力(K d = 15±1 μM)与ATP与已报道的适体的结合亲和力相比。分子动力学模拟支持了突变体的这些特征。

































 京公网安备 11010802027423号
京公网安备 11010802027423号