Bioorganic Chemistry ( IF 4.5 ) Pub Date : 2018-08-28 , DOI: 10.1016/j.bioorg.2018.08.037
Mamali Das , Sengodu Prakash , Chirasmita Nayak , Nandhini Thangavel , Sanjeev Kumar Singh , Paramasivam Manisankar , Kasi Pandima Devi
|
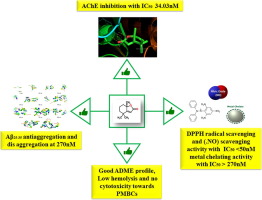
Synthesis of natural products has speeded up drug discovery process by minimizing the time for their purification from natural source. Several diseases like Alzheimer's disease (AD) demand exploring multi targeted drug candidates, and for the first time we report the multi AD target inhibitory potential of synthesized dihydroactinidiolide (DA). Though the activity of DA in several solvent extracts have been proved to possess free radical scavenging, anti bacterial and anti cancer activities, its neuroprotective efficacy has not been evidenced yet. Hence DA was successfully synthesized from β-ionone using facile two-step oxidation method. It showed potent acetylcholinesterase (AChE) inhibition with half maximal inhibitory concentration (IC50) 34.03 nM, which was further supported by molecular docking results showing strong H bonding with some of the active site residues such as GLY117, GLY119 and SER200 of AChE. Further it displayed DPPH and (.NO) scavenging activity with IC50 value 50 nM and metal chelating activity with IC50 >270 nM. Besides, it significantly prevented amyloid β25-35 self-aggregation and promoted its disaggregation at 270 nM. It did not show cytotoxic effect towards Neuro2a (N2a) cells up to 24 h at 50 and 270 nM while it significantly increased viability of amyloid β25-35 treated N2a cells through ROS generation at both the concentrations. Cytotoxicity profile of DA against human PBMC was quite impressive. Hemolysis studies also revealed very low hemolysis i.e. minimum 2.35 to maximum 5.61%. It also had suitable ADME properties which proved its druglikeness. The current findings demand for further in vitro and in vivo studies to develop DA as a multi target lead against AD.
中文翻译:
二氢actinidiolide,一种抗Aβ25-35诱导的Neuro2a细胞毒性的天然产物:合成,计算机模拟和体外研究
天然产物的合成通过最大程度地减少从天然来源纯化的时间而加快了药物开发过程。像阿尔茨海默氏病(AD)在内的几种疾病都需要探索多种靶向药物候选物,并且我们首次报道了合成的二氢肌醇(DA)的多种AD靶标抑制潜能。尽管已经证明DA在几种溶剂提取物中的活性具有自由基清除,抗细菌和抗癌活性,但尚未证明其神经保护功效。因此,采用简便的两步氧化法成功地由β-紫罗兰酮合成了DA。它显示出有效的乙酰胆碱酯酶(AChE)抑制作用,最大抑制浓度为一半(IC 50)34.03 nM,这进一步得到分子对接结果的支持,显示出与某些活性位点残基如AChE的GLY117,GLY119和SER200的强H键合。此外,它还显示出DPPH和(.NO)清除活性,IC 50值为50 nM,金属螯合活性为IC 50 > 270 nM。此外,它在270 nM时可显着阻止淀粉样蛋白β25-35的自聚集并促进其解聚。它在50和270 nM的作用下长达24 h均未显示对Neuro2a(N2a)细胞的细胞毒性作用,而显着增加了淀粉样蛋白25-35的活力两种浓度下通过ROS生成的RNA处理了N2a细胞。DA对人PBMC的细胞毒性特征令人印象深刻。溶血研究还显示溶血非常低,即最低2.35至最高5.61%。它还具有合适的ADME性质,证明了它的药物相似性。目前的发现要求进一步的体外和体内研究以将DA发展为对抗AD的多靶标。
































 京公网安备 11010802027423号
京公网安备 11010802027423号