当前位置:
X-MOL 学术
›
Am. J. Hum. Genet.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A One-Penny Imputed Genome from Next-Generation Reference Panels.
American Journal of Human Genetics ( IF 8.1 ) Pub Date : 2018-08-09 , DOI: 10.1016/j.ajhg.2018.07.015 Brian L Browning 1 , Ying Zhou 2 , Sharon R Browning 2
American Journal of Human Genetics ( IF 8.1 ) Pub Date : 2018-08-09 , DOI: 10.1016/j.ajhg.2018.07.015 Brian L Browning 1 , Ying Zhou 2 , Sharon R Browning 2
Affiliation
|
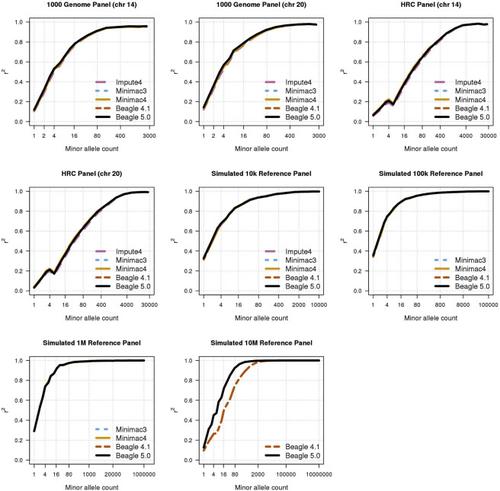
Genotype imputation is commonly performed in genome-wide association studies because it greatly increases the number of markers that can be tested for association with a trait. In general, one should perform genotype imputation using the largest reference panel that is available because the number of accurately imputed variants increases with reference panel size. However, one impediment to using larger reference panels is the increased computational cost of imputation. We present a new genotype imputation method, Beagle 5.0, which greatly reduces the computational cost of imputation from large reference panels. We compare Beagle 5.0 with Beagle 4.1, Impute4, Minimac3, and Minimac4 using 1000 Genomes Project data, Haplotype Reference Consortium data, and simulated data for 10k, 100k, 1M, and 10M reference samples. All methods produce nearly identical accuracy, but Beagle 5.0 has the lowest computation time and the best scaling of computation time with increasing reference panel size. For 10k, 100k, 1M, and 10M reference samples and 1,000 phased target samples, Beagle 5.0’s computation time is 3× (10k), 12× (100k), 43× (1M), and 533× (10M) faster than the fastest alternative method. Cost data from the Amazon Elastic Compute Cloud show that Beagle 5.0 can perform genome-wide imputation from 10M reference samples into 1,000 phased target samples at a cost of less than one US cent per sample.
中文翻译:

来自下一代参考小组的一分钱估算基因组。
基因型插补通常在全基因组关联研究中进行,因为它大大增加了可以测试与性状关联的标志物的数量。通常,应该使用可用的最大参考面板进行基因型插补,因为精确插补的变体的数量会随着参考面板尺寸的增加而增加。但是,使用较大的参考面板的一个障碍是估算的计算成本增加。我们提出了一种新的基因型插补方法Beagle 5.0,该方法大大降低了从大型参考面板进行插补的计算成本。我们使用1000个基因组计划数据,Haplotype Reference Consortium数据以及10k,100k,1M和10M参考样本的模拟数据,将Beagle 5.0与Beagle 4.1,Imute4,Minimac3和Minimac4进行了比较。所有方法产生的精度几乎相同,但是Beagle 5.0随着参考面板尺寸的增加,具有最短的计算时间和最佳的计算时间缩放比例。对于10k,100k,1M和10M参考样本以及1,000个定相目标样本,Beagle 5.0的计算时间比以下分别快3倍(10k),12倍(100k),43倍(1M)和533倍(10M)。最快的替代方法。来自Amazon Elastic Compute Cloud的成本数据显示,Beagle 5.0可以从1000万个参考样本到1000个分阶段的目标样本中进行全基因组估算,而每个样本的成本不到一美分。比最快的替代方法快43倍(1M)和533倍(10M)。来自Amazon Elastic Compute Cloud的成本数据显示,Beagle 5.0可以从1000万个参考样本到1000个分阶段的目标样本中进行全基因组估算,而每个样本的成本不到一美分。比最快的替代方法快43倍(1M)和533倍(10M)。来自Amazon Elastic Compute Cloud的成本数据显示,Beagle 5.0可以从1000万个参考样本到1000个分阶段的目标样本中进行全基因组估算,而每个样本的成本不到一美分。
更新日期:2018-08-17
中文翻译:
来自下一代参考小组的一分钱估算基因组。
基因型插补通常在全基因组关联研究中进行,因为它大大增加了可以测试与性状关联的标志物的数量。通常,应该使用可用的最大参考面板进行基因型插补,因为精确插补的变体的数量会随着参考面板尺寸的增加而增加。但是,使用较大的参考面板的一个障碍是估算的计算成本增加。我们提出了一种新的基因型插补方法Beagle 5.0,该方法大大降低了从大型参考面板进行插补的计算成本。我们使用1000个基因组计划数据,Haplotype Reference Consortium数据以及10k,100k,1M和10M参考样本的模拟数据,将Beagle 5.0与Beagle 4.1,Imute4,Minimac3和Minimac4进行了比较。所有方法产生的精度几乎相同,但是Beagle 5.0随着参考面板尺寸的增加,具有最短的计算时间和最佳的计算时间缩放比例。对于10k,100k,1M和10M参考样本以及1,000个定相目标样本,Beagle 5.0的计算时间比以下分别快3倍(10k),12倍(100k),43倍(1M)和533倍(10M)。最快的替代方法。来自Amazon Elastic Compute Cloud的成本数据显示,Beagle 5.0可以从1000万个参考样本到1000个分阶段的目标样本中进行全基因组估算,而每个样本的成本不到一美分。比最快的替代方法快43倍(1M)和533倍(10M)。来自Amazon Elastic Compute Cloud的成本数据显示,Beagle 5.0可以从1000万个参考样本到1000个分阶段的目标样本中进行全基因组估算,而每个样本的成本不到一美分。比最快的替代方法快43倍(1M)和533倍(10M)。来自Amazon Elastic Compute Cloud的成本数据显示,Beagle 5.0可以从1000万个参考样本到1000个分阶段的目标样本中进行全基因组估算,而每个样本的成本不到一美分。






























 京公网安备 11010802027423号
京公网安备 11010802027423号