当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Total Syntheses of Thailanstatins A–C, Spliceostatin D, and Analogues Thereof. Stereodivergent Synthesis of Tetrasubstituted Dihydro- and Tetrahydropyrans and Design, Synthesis, Biological Evaluation, and Discovery of Potent Antitumor Agents
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2018-06-26 , DOI: 10.1021/jacs.8b04634 K. C. Nicolaou 1 , Derek Rhoades 1 , S. Mothish Kumar 1
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2018-06-26 , DOI: 10.1021/jacs.8b04634 K. C. Nicolaou 1 , Derek Rhoades 1 , S. Mothish Kumar 1
Affiliation
|
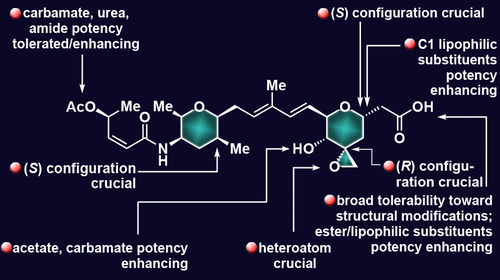
Efficient and selective total syntheses of spliceosome modulating natural products thailanstatins A-C and spliceostatin D are reported. A number of stereoselective methods for the construction of various tetrasubstituted dihydro- and tetrahydropyrans were developed as a prerequisite for the syntheses of these naturally occurring molecules and variations thereof. The pyran-forming reactions utilize a Heck/Saegusa-Ito cascade sequence to generate hydroxy α,β,γ,δ-unsaturated aldehyde precursors followed by a catalyst-controlled oxa-Michael cyclization to furnish tetrasubstituted dihydropyrans with high stereocontrol. Subsequent optimized homogeneous or heterogeneous hydrogenations of these dihydropyran systems afford their tetrahydropyran counterparts, also in a highly stereoselective manner. The synthesized thailanstatins and related analogues were biologically evaluated for their cytotoxic properties, leading to the identification of a number of compounds with exceptionally potent antitumor activities suitable for further development as potential antibody-drug conjugate payloads, single drugs, or drug combinations for cancer therapies. Important structure-activity relationships within the thailanstatin family and structurally related compounds are discussed and are expected to be path-pointing for future studies.
中文翻译:

Thailanstatin A-C、Spliceostatin D 及其类似物的全合成。四取代二氢和四氢吡喃的立体发散合成以及有效抗肿瘤剂的设计、合成、生物学评价和发现
报道了剪接体调节天然产物泰兰他汀 AC 和剪接抑制素 D 的高效和选择性全合成。开发了许多用于构建各种四取代二氢和四氢吡喃的立体选择性方法,作为合成这些天然存在的分子及其变体的先决条件。吡喃形成反应利用 Heck/Saegusa-Ito 级联序列生成羟基 α,β,γ,δ-不饱和醛前体,然后通过催化剂控制的 oxa-Michael 环化提供具有高度立体控制的四取代二氢吡喃。随后对这些二氢吡喃系统进行优化的均相或非均相氢化,也以高度立体选择性的方式提供了它们的四氢吡喃对应物。合成的泰兰他汀和相关类似物对其细胞毒性特性进行了生物学评估,从而鉴定了许多具有异常有效抗肿瘤活性的化合物,适合进一步开发为潜在的抗体药物偶联物有效载荷、单一药物或用于癌症治疗的药物组合。讨论了泰兰他汀家族中重要的构效关系和结构相关的化合物,并有望成为未来研究的方向。
更新日期:2018-06-26
中文翻译:
Thailanstatin A-C、Spliceostatin D 及其类似物的全合成。四取代二氢和四氢吡喃的立体发散合成以及有效抗肿瘤剂的设计、合成、生物学评价和发现
报道了剪接体调节天然产物泰兰他汀 AC 和剪接抑制素 D 的高效和选择性全合成。开发了许多用于构建各种四取代二氢和四氢吡喃的立体选择性方法,作为合成这些天然存在的分子及其变体的先决条件。吡喃形成反应利用 Heck/Saegusa-Ito 级联序列生成羟基 α,β,γ,δ-不饱和醛前体,然后通过催化剂控制的 oxa-Michael 环化提供具有高度立体控制的四取代二氢吡喃。随后对这些二氢吡喃系统进行优化的均相或非均相氢化,也以高度立体选择性的方式提供了它们的四氢吡喃对应物。合成的泰兰他汀和相关类似物对其细胞毒性特性进行了生物学评估,从而鉴定了许多具有异常有效抗肿瘤活性的化合物,适合进一步开发为潜在的抗体药物偶联物有效载荷、单一药物或用于癌症治疗的药物组合。讨论了泰兰他汀家族中重要的构效关系和结构相关的化合物,并有望成为未来研究的方向。










































 京公网安备 11010802027423号
京公网安备 11010802027423号