Polymer ( IF 4.1 ) Pub Date : 2018-06-18 , DOI: 10.1016/j.polymer.2018.06.049 Yanping Ma , Chenchen Hu , Hongxia Guo , Lin Fan , Shiyong Yang , Wen-Hua Sun
|
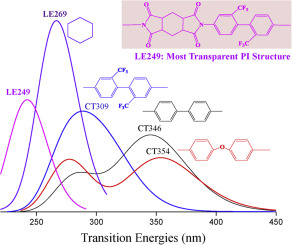
Understanding of the nature of the transition mechanism in UV-visible absorption spectrum may contribute to the designing of highly transparent polyimide (PI) materials. In the present work, structure effect (alicyclic ring, flexible -O- linkage, and -CF3) on the intra- and inter-molecular transition mechanism of optical absorptions in PI model molecules is investigated by the Density functional theory (DFT) in combination with the time dependent DFT calculations. Six pyromelliticdianhydride (PMDA) derived aromatic and six 1,2,4,5-cyclohexanetetracarboxylic dianhydride (CHDA) derived semi-aromatic polyimide (PI) species are considered and their intermolecular transitions are analyzed by designing typical dimers with classical face-to-face π-π stacking orientation. The natural bond orbital method (NBO) is employed to analyze the delocalization degree in the PI species. By calculating the energies of the HOMO (highest occupied molecular orbital) and LUMO (lowest unoccupied molecular orbital) as well as the transition characters between them, the electron excitation nature concerning on the structural variations in the monomers and the dimers are clarified. These theoretical results agree well with the related experimental observations and provide a deep understanding into the transition mechanism of optical absorptions in the PI species.
中文翻译:
聚酰亚胺中紫外可见吸收光谱跃迁机理的结构效应:密度泛函理论研究
了解紫外线-可见光吸收光谱中跃迁机理的性质可能有助于设计高度透明的聚酰亚胺(PI)材料。另外,在本工作中,结构效应(脂环,柔性-O-键,和-CF 3的)帧内和帧间通过密度泛函理论(DFT)结合时间相关DFT计算,研究了PI模型分子中光吸收的分子跃迁机理。考虑了六个均苯四酸二酐(PMDA)衍生的芳族化合物和六个1,2,4,5-环己烷四甲酸二酐(CHDA)衍生的半芳族聚酰亚胺(PI)物种,并通过设计经典面对面的典型二聚体来分析它们的分子间转变。 π-π堆叠方向。采用自然键轨道法(NBO)分析PI物种的离域度。通过计算HOMO(最高占据分子轨道)和LUMO(最低未占据分子轨道)的能量以及它们之间的跃迁特性,阐明了与单体和二聚体的结构变化有关的电子激发性质。这些理论结果与相关的实验观察非常吻合,并提供了对PI物种中光吸收跃迁机制的深入理解。










































 京公网安备 11010802027423号
京公网安备 11010802027423号