当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Theoretical Insight into Catalytic Propane Dehydrogenation on Ni(111)
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2018-06-15 , DOI: 10.1021/acs.jpcc.8b03939
Tinnakorn Saelee 1 , Supawadee Namuangruk 2 , Nawee Kungwan 1, 3 , Anchalee Junkaew 2
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2018-06-15 , DOI: 10.1021/acs.jpcc.8b03939
Tinnakorn Saelee 1 , Supawadee Namuangruk 2 , Nawee Kungwan 1, 3 , Anchalee Junkaew 2
Affiliation
|
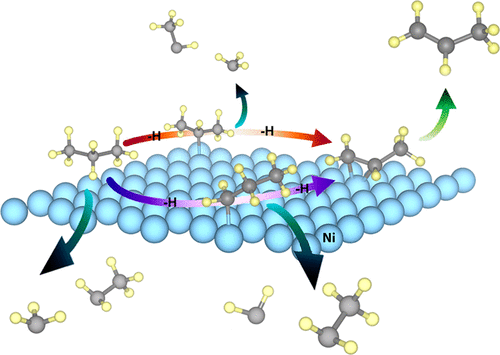
Here, propane dehydrogenation (PDH) to propylene and side reactions, namely, cracking and deep dehydrogenation on Ni(111) surface, have been theoretically investigated by density functional theory calculation. On the basis of adsorption energies, propane is physisorbed on Ni(111) surface, whereas propylene exhibits chemisorption supported by electronic charge results. In the PDH reaction, possible pathways can occur via two possible intermediates, i.e., 1-propyl and 2-propyl. Our results suggest that PDH reaction through 1-propyl intermediate is both kinetically and thermodynamically more favorable than another pathway. The C–C bond cracking during PDH process is more difficult to occur than the C–H activation reaction because of higher energy barrier of the C–C bond cracking. However, deep dehydrogenation is the preferable process after PDH, owing to the strong adsorption of propylene on Ni(111) surface, resulting in low selectivity of propylene production. This work suggests that Ni(111) has superior activity toward PDH; however, the enhancement of propylene desorption is required to improve its selectivity. The understanding in molecular level from this work is useful for designing and developing better Ni-based catalysts in terms of activity and selectivity for propane conversion to propylene.
中文翻译:

Ni(111)催化丙烷脱氢的理论研究
在这里,通过密度泛函理论计算,对丙烷脱氢(PDH)生成丙烯和副反应(即Ni(111)表面的裂解和深度脱氢)进行了理论研究。根据吸附能,丙烷物理吸附在Ni(111)表面,而丙烯则表现出化学吸附,这是由电荷结果支持的。在PDH反应中,可能的途径可以通过两种可能的中间体,即1-丙基和2-丙基发生。我们的结果表明,通过1-丙基中间体进行的PDH反应在动力学和热力学上均比其他途径更有利。PDH过程中的C–C键断裂比C–H活化反应更难发生,因为C–C键断裂的能垒更高。但是,深度脱氢是PDH之后的首选工艺,由于丙烯在Ni(111)表面上的强烈吸附,导致丙烯生产的选择性低。这项工作表明Ni(111)对PDH具有更好的活性。但是,需要提高丙烯的解吸能力以提高其选择性。从这项工作的分子水平上的理解,对于丙烷转化为丙烯的活性和选择性方面的设计和开发更好的镍基催化剂很有用。
更新日期:2018-06-15
中文翻译:
Ni(111)催化丙烷脱氢的理论研究
在这里,通过密度泛函理论计算,对丙烷脱氢(PDH)生成丙烯和副反应(即Ni(111)表面的裂解和深度脱氢)进行了理论研究。根据吸附能,丙烷物理吸附在Ni(111)表面,而丙烯则表现出化学吸附,这是由电荷结果支持的。在PDH反应中,可能的途径可以通过两种可能的中间体,即1-丙基和2-丙基发生。我们的结果表明,通过1-丙基中间体进行的PDH反应在动力学和热力学上均比其他途径更有利。PDH过程中的C–C键断裂比C–H活化反应更难发生,因为C–C键断裂的能垒更高。但是,深度脱氢是PDH之后的首选工艺,由于丙烯在Ni(111)表面上的强烈吸附,导致丙烯生产的选择性低。这项工作表明Ni(111)对PDH具有更好的活性。但是,需要提高丙烯的解吸能力以提高其选择性。从这项工作的分子水平上的理解,对于丙烷转化为丙烯的活性和选择性方面的设计和开发更好的镍基催化剂很有用。
































 京公网安备 11010802027423号
京公网安备 11010802027423号