当前位置:
X-MOL 学术
›
J. Chem. Phys.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Spin-flip, tensor equation-of-motion configuration interaction with a density-functional correction: A spin-complete method for exploring excited-state potential energy surfaces
The Journal of Chemical Physics ( IF 3.1 ) Pub Date : 2015-12-17 14:41:21 , DOI: 10.1063/1.4937571 Xing Zhang 1 , John M. Herbert 1
The Journal of Chemical Physics ( IF 3.1 ) Pub Date : 2015-12-17 14:41:21 , DOI: 10.1063/1.4937571 Xing Zhang 1 , John M. Herbert 1
Affiliation
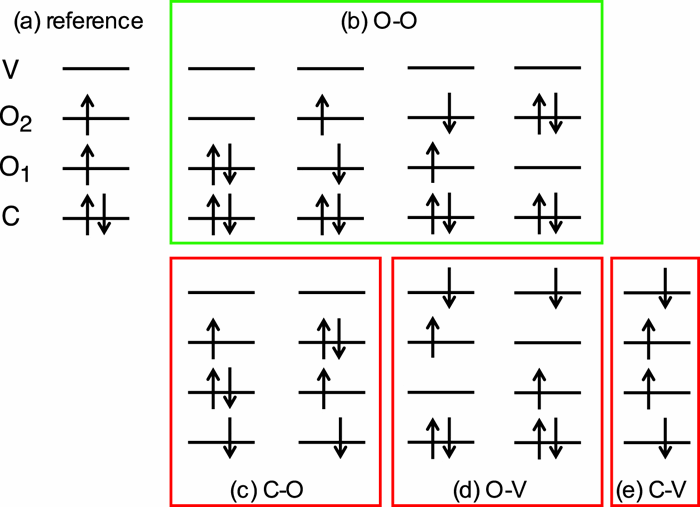
|
We revisit the formalism of the spin-adapted, spin-flip (SA-SF) configuration-interaction singles (CIS) method based on a tensor equation-of-motion formalism that affords proper spin eigenstates without sacrificing single-reference simplicity. Matrix elements for SA-SF-CIS are then modified in a manner similar to collinear spin-flip time-dependent density functional theory (SF-TDDFT), to include a DFT exchange-correlation correction. The performance of this method, which we call SA-SF-DFT, is evaluated numerically and we find that it systematically improves the energies of electronic states that exhibit significant spin contamination within the conventional SF-TDDFT approach. The new method cures the state assignment problem that plagues geometry optimizations and ab initiomolecular dynamics simulations using traditional SF-TDDFT, without sacrificing computational efficiency, and furthermore provides correct topology at conical intersections, including those that involve the ground state, unlike conventional TDDFT. As such, SA-SF-DFT appears to be a promising method for generating excited-state potential energy surfaces at DFT cost.
中文翻译:

自旋翻转,张量运动方程与密度函数校正的相互作用:一种探索激发态势能面的自旋完全方法
我们基于张量运动方程式的形式主义重新审视了自旋适应的自旋翻转(SA-SF)配置相互作用单(CIS)方法的形式主义,该方法提供了适当的自旋本征态而又不牺牲单一参考的简单性。然后以类似于共线自旋翻转时间相关密度泛函理论(SF-TDDFT)的方式修改SA-SF-CIS的矩阵元素,以包括DFT交换相关校正。对该方法的性能(我们称为SA-SF-DFT)进行了数值评估,我们发现它可以系统地提高在常规SF-TDDFT方法中表现出明显自旋污染的电子态的能量。新方法解决了困扰几何优化和从头算起的状态分配问题使用传统的SF-TDDFT进行分子动力学模拟,而不会牺牲计算效率,而且与传统的TDDFT不同,它在圆锥形交叉点(包括那些涉及基态的交叉点)提供了正确的拓扑。这样,SA-SF-DFT似乎是一种以DFT成本产生激发态势能面的有前途的方法。
更新日期:2015-12-18
中文翻译:
自旋翻转,张量运动方程与密度函数校正的相互作用:一种探索激发态势能面的自旋完全方法
我们基于张量运动方程式的形式主义重新审视了自旋适应的自旋翻转(SA-SF)配置相互作用单(CIS)方法的形式主义,该方法提供了适当的自旋本征态而又不牺牲单一参考的简单性。然后以类似于共线自旋翻转时间相关密度泛函理论(SF-TDDFT)的方式修改SA-SF-CIS的矩阵元素,以包括DFT交换相关校正。对该方法的性能(我们称为SA-SF-DFT)进行了数值评估,我们发现它可以系统地提高在常规SF-TDDFT方法中表现出明显自旋污染的电子态的能量。新方法解决了困扰几何优化和从头算起的状态分配问题使用传统的SF-TDDFT进行分子动力学模拟,而不会牺牲计算效率,而且与传统的TDDFT不同,它在圆锥形交叉点(包括那些涉及基态的交叉点)提供了正确的拓扑。这样,SA-SF-DFT似乎是一种以DFT成本产生激发态势能面的有前途的方法。






























 京公网安备 11010802027423号
京公网安备 11010802027423号