当前位置:
X-MOL 学术
›
Org. Biomol. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
An ab initio and DFT study of trifluoromethylation using Umemoto's reagent†
Organic & Biomolecular Chemistry ( IF 2.9 ) Pub Date : 2018-05-30 00:00:00 , DOI: 10.1039/c8ob00805a Satoko Tsujibayashi 1, 2, 3, 4, 5 , Yutaka Kataoka 1, 2, 3, 4, 5 , Shun Hirano 1, 2, 3, 4, 5 , Hiroshi Matsubara 1, 2, 3, 4, 5
Organic & Biomolecular Chemistry ( IF 2.9 ) Pub Date : 2018-05-30 00:00:00 , DOI: 10.1039/c8ob00805a Satoko Tsujibayashi 1, 2, 3, 4, 5 , Yutaka Kataoka 1, 2, 3, 4, 5 , Shun Hirano 1, 2, 3, 4, 5 , Hiroshi Matsubara 1, 2, 3, 4, 5
Affiliation
|
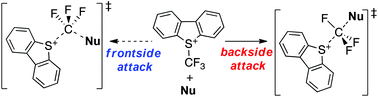
Trifluoromethylation using Umemoto's reagent is an important transformation that allows the preparation of compounds bearing trifluoromethyl groups. To investigate the mechanism of this reaction, ab initio and density functional theory (DFT) calculations were carried out using pyrrole, aniline, sodium acetylacetonate, and sodium methyl acetoacetate as nucleophiles. At the highest level of theory examined (i.e., CCSD(T)/6-311+G(d,p)//M06-2X/6-311+G(d,p)), the energy barriers for the forward process (ΔE‡1) of both the backside and frontside attack of pyrrole on a model Umemoto reagent (i.e., S-(trifluoromethyl)dimethylsulfonium, CF3DMS) were predicted to be 135.9 and 192.3 kJ mol−1, respectively, while values of 131.9 and 188.2 kJ mol−1 were obtained at the MP2/6-311+G(d,p)//M06-2X/6-31+G(d,p) level. These outcomes suggest that the reaction proceeds via the backside mechanism. Using the MP2 method, the investigation of the trifluoromethylation of pyrrole and sodium acetoacetate with the sulfonium moiety of Umemoto's reagent, S-(trifluoromethyl)dibenzothionium, revealed that this reaction would also occur through the backside mechanism, thereby indicating that this pathway remains feasible despite solvent effects. Finally, computational investigations revealed that the simple single-electron transfer mechanism, which should occur between Umemoto's reagent and nucleophiles, did not take place during this reaction.
中文翻译:

一个从头开始,用梅本试剂三氟甲基化的DFT研究†
使用梅本氏试剂进行的三氟甲基化是一种重要的转化方法,可以制备带有三氟甲基的化合物。为了研究该反应的机理,使用吡咯,苯胺,乙酰丙酮钠和乙酰乙酸甲酯钠作为亲核试剂进行了从头算和密度泛函理论(DFT)的计算。在最高理论水平下(即CCSD(T)/ 6-311 + G(d,p)// M06-2X / 6-311 + G(d,p)),正向过程的能量壁垒(Δ ë ‡ 1)的背面和吡咯上的模型正面攻击两者的梅本试剂(即,小号- (三氟甲基)二甲基,CF 3DMS)预计分别为135.9和192.3 kJ mol -1,而在MP2 / 6-311 + G(d,p)// M06-2X / 6-处获得的值为131.9和188.2 kJ mol -1。 31 + G(d,p)等级。这些结果表明反应是通过背面机制进行的。使用MP2方法研究吡咯和乙酰乙酸钠与梅本试剂S的sulf部分的三氟甲基化-(三氟甲基)二苯并硫鎓,表明该反应也将通过背面机理发生,从而表明尽管有溶剂作用,该途径仍然可行。最后,计算研究表明,在梅本试剂和亲核试剂之间应该发生的简单的单电子转移机制并未在该反应过程中发生。
更新日期:2018-05-30
中文翻译:
一个从头开始,用梅本试剂三氟甲基化的DFT研究†
使用梅本氏试剂进行的三氟甲基化是一种重要的转化方法,可以制备带有三氟甲基的化合物。为了研究该反应的机理,使用吡咯,苯胺,乙酰丙酮钠和乙酰乙酸甲酯钠作为亲核试剂进行了从头算和密度泛函理论(DFT)的计算。在最高理论水平下(即CCSD(T)/ 6-311 + G(d,p)// M06-2X / 6-311 + G(d,p)),正向过程的能量壁垒(Δ ë ‡ 1)的背面和吡咯上的模型正面攻击两者的梅本试剂(即,小号- (三氟甲基)二甲基,CF 3DMS)预计分别为135.9和192.3 kJ mol -1,而在MP2 / 6-311 + G(d,p)// M06-2X / 6-处获得的值为131.9和188.2 kJ mol -1。 31 + G(d,p)等级。这些结果表明反应是通过背面机制进行的。使用MP2方法研究吡咯和乙酰乙酸钠与梅本试剂S的sulf部分的三氟甲基化-(三氟甲基)二苯并硫鎓,表明该反应也将通过背面机理发生,从而表明尽管有溶剂作用,该途径仍然可行。最后,计算研究表明,在梅本试剂和亲核试剂之间应该发生的简单的单电子转移机制并未在该反应过程中发生。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号