当前位置:
X-MOL 学术
›
ACS Appl. Mater. Interfaces
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Ab Initio Metadynamics Study of the VO2+/VO2+ Redox Reaction Mechanism at the Graphite Edge/Water Interface
ACS Applied Materials & Interfaces ( IF 8.3 ) Pub Date : 2018-05-29 00:00:00 , DOI: 10.1021/acsami.8b05864 Zhen Jiang , Konstantin Klyukin , Vitaly Alexandrov
ACS Applied Materials & Interfaces ( IF 8.3 ) Pub Date : 2018-05-29 00:00:00 , DOI: 10.1021/acsami.8b05864 Zhen Jiang , Konstantin Klyukin , Vitaly Alexandrov
|
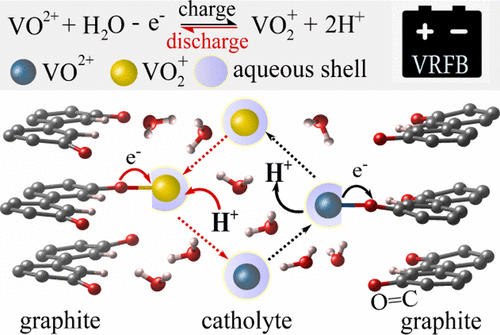
Redox flow batteries (RFBs) are promising electrochemical energy storage systems, for which development is impeded by a poor understanding of redox reactions occurring at electrode/electrolyte interfaces. Even for the conventional all-vanadium RFB chemistry employing V2+/V3+ and VO2+/VO2+ couples, there is still no consensus about the reaction mechanism, electrode active sites, and rate-determining step. Herein, we perform Car–Parrinello molecular dynamics-based metadynamics simulations to unravel the mechanism of the VO2+/VO2+ redox reaction in water at the oxygen-functionalized graphite (112̅0) edge surface serving as a representative carbon-based electrode. Our results suggest that during the battery discharge aqueous VO2+/VO2+ species adsorb at the surface C–O groups as inner-sphere complexes, exhibiting faster adsorption/desorption kinetics than V2+/V3+, at least at low vanadium concentrations considered in our study. We find that this is because (i) VO2+/VO2+ conversion does not involve the slow transfer of an oxygen atom, (ii) protonation of VO2+ is spontaneous and coupled to interfacial electron transfer in acidic conditions to enable VO2+ formation, and (iii) V3+ found to be strongly bound to oxygen groups of the graphite surface features unfavorable desorption kinetics. In contrast, the reverse process taking place upon charging is expected to be more sluggish for the VO2+/VO2+ redox couple because of both unfavorable deprotonation of the VO2+ water ligands and adsorption/desorption kinetics.
中文翻译:

石墨边缘/水界面VO 2 + / VO 2+氧化还原反应机理的从头算动力学研究
氧化还原液流电池(RFB)是有前途的电化学储能系统,由于对电极/电解质界面处发生的氧化还原反应的了解不足,阻碍了其发展。即使对于使用V 2+ / V 3+和VO 2 + / VO 2+对的常规全钒RFB化学方法,在反应机理,电极活性位点和速率确定步骤方面仍未达成共识。在这里,我们执行基于汽车-帕里内罗分子动力学的元动力学模拟,以揭示VO 2 + / VO 2+的机理氧官能化的石墨(112̅0)边缘表面上的水氧化还原反应,是代表性的碳基电极。我们的结果表明,在电池放电期间,VO 2 + / VO 2+物质作为内球络合物在表面C–O基团上吸附,比V 2+ / V 3+表现出更快的吸附/解吸动力学,至少在低温下我们研究中考虑的钒浓度。我们发现这是因为(i)VO 2 + / VO 2+的转化不涉及氧原子的缓慢转移,(ii)VO 2 +的质子化是自发的,并在酸性条件下与界面电子转移耦合,以使VO 2+形成,并且(iii)被发现与石墨表面的氧基团牢固结合的V 3+具有不利的解吸动力学。相反,由于VO 2+水配体的不利去质子化和吸附/解吸动力学,对于VO 2 + / VO 2+氧化还原对,预期在充电时发生的逆过程将更缓慢。
更新日期:2018-05-29
中文翻译:
石墨边缘/水界面VO 2 + / VO 2+氧化还原反应机理的从头算动力学研究
氧化还原液流电池(RFB)是有前途的电化学储能系统,由于对电极/电解质界面处发生的氧化还原反应的了解不足,阻碍了其发展。即使对于使用V 2+ / V 3+和VO 2 + / VO 2+对的常规全钒RFB化学方法,在反应机理,电极活性位点和速率确定步骤方面仍未达成共识。在这里,我们执行基于汽车-帕里内罗分子动力学的元动力学模拟,以揭示VO 2 + / VO 2+的机理氧官能化的石墨(112̅0)边缘表面上的水氧化还原反应,是代表性的碳基电极。我们的结果表明,在电池放电期间,VO 2 + / VO 2+物质作为内球络合物在表面C–O基团上吸附,比V 2+ / V 3+表现出更快的吸附/解吸动力学,至少在低温下我们研究中考虑的钒浓度。我们发现这是因为(i)VO 2 + / VO 2+的转化不涉及氧原子的缓慢转移,(ii)VO 2 +的质子化是自发的,并在酸性条件下与界面电子转移耦合,以使VO 2+形成,并且(iii)被发现与石墨表面的氧基团牢固结合的V 3+具有不利的解吸动力学。相反,由于VO 2+水配体的不利去质子化和吸附/解吸动力学,对于VO 2 + / VO 2+氧化还原对,预期在充电时发生的逆过程将更缓慢。





























 京公网安备 11010802027423号
京公网安备 11010802027423号