当前位置:
X-MOL 学术
›
Arch. Pharm.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Design, synthesis, in vitro and in silico evaluation of new pyrrole derivatives as monoamine oxidase inhibitors
Archiv der Pharmazie ( IF 4.3 ) Pub Date : 2018-05-22 , DOI: 10.1002/ardp.201800082 Mehlika D. Altintop 1 , Belgin Sever 1 , Derya Osmaniye 1 , Begüm N. Sağlık 1 , Ahmet Özdemir 1
Archiv der Pharmazie ( IF 4.3 ) Pub Date : 2018-05-22 , DOI: 10.1002/ardp.201800082 Mehlika D. Altintop 1 , Belgin Sever 1 , Derya Osmaniye 1 , Begüm N. Sağlık 1 , Ahmet Özdemir 1
Affiliation
|
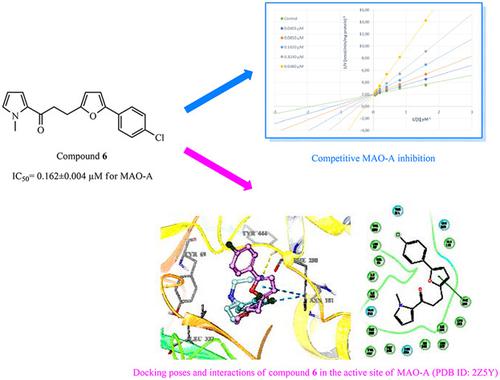
In an effort to develop potent monoamine oxidase (MAO) inhibitors, new pyrrole derivatives were obtained via the selective reduction of the CC bonds of 1‐(1‐methyl‐1H‐pyrrol‐2‐yl)‐3‐[5‐(aryl)furan‐2‐yl]prop‐2‐en‐1‐ones through palladium catalyzed hydrogenation in ethanol. The synthesized compounds were screened for their inhibitory effects on MAO‐A and MAO‐B by an in vitro fluorometric method. The selectivity index (SI) value was given as the ratio of IC50 (MAO‐A)/IC50 (MAO‐B) for each compound. 3‐(5‐(4‐Chlorophenyl)furan‐2‐yl)‐1‐(1‐methyl‐1H‐pyrrol‐2‐yl)propan‐1‐one (6) was identified as the most selective MAO‐A inhibitor in this series, with an IC50 value of 0.162 µM and a SI value of 0.002. Kinetic studies were also carried out to assess the nature of MAO‐A inhibition by compound 6. According to Lineweaver–Burk plots, compound 6 was found to be a competitive MAO‐A inhibitor and the Ki value of compound 6 was determined as 0.1221 μM. Docking studies were performed for compound 6 and clorgyline using the human MAO‐A crystal structure (PDB ID: 2Z5Y). The docking results showed that compound 6 presented similar interactions as clorgyline in the active center cavity of the enzyme. Molinspiration software was used to determine the physicochemical parameters of all compounds for an evaluation of their compliance to Lipinski's rule of five. Compound 6 did not violate Lipinski's rule, making it a potential orally bioavailable therapeutic agent.
中文翻译:

作为单胺氧化酶抑制剂的新型吡咯衍生物的设计、合成、体外和计算机评估
为了开发有效的单胺氧化酶 (MAO) 抑制剂,通过选择性还原 1-(1-甲基-1H-吡咯-2-基)-3-[5- (芳基)呋喃-2-基]丙-2-烯-1-酮通过钯催化在乙醇中加氢。通过体外荧光法筛选合成的化合物对 MAO-A 和 MAO-B 的抑制作用。选择性指数 (SI) 值以每种化合物的 IC50 (MAO-A)/IC50 (MAO-B) 的比率给出。3-(5-(4-Chlorophenyl)furan-2-yl)-1-(1-methyl-1H-pyrrol-2-yl)propan-1-one (6) 被鉴定为最具选择性的 MAO-A 抑制剂在该系列中,IC50 值为 0.162 µM,SI 值为 0.002。还进行了动力学研究以评估化合物 6 对 MAO-A 抑制的性质。 根据 Lineweaver-Burk 图,发现化合物 6 是一种竞争性 MAO-A 抑制剂,化合物 6 的 Ki 值确定为 0.1221 μM。使用人 MAO-A 晶体结构(PDB ID:2Z5Y)对化合物 6 和氯吉林进行对接研究。对接结果表明,化合物6在酶的活性中心腔中表现出与氯吉林相似的相互作用。Molinspiration 软件用于确定所有化合物的理化参数,以评估它们是否符合 Lipinski 的五法则。化合物 6 没有违反 Lipinski 规则,使其成为潜在的口服生物可利用治疗剂。对接结果表明,化合物6在酶的活性中心腔中表现出与氯吉林相似的相互作用。Molinspiration 软件用于确定所有化合物的理化参数,以评估它们是否符合 Lipinski 的五法则。化合物 6 没有违反 Lipinski 规则,使其成为潜在的口服生物可利用治疗剂。对接结果表明,化合物6在酶的活性中心腔中表现出与氯吉林相似的相互作用。Molinspiration 软件用于确定所有化合物的理化参数,以评估它们是否符合 Lipinski 的五法则。化合物 6 没有违反 Lipinski 规则,使其成为潜在的口服生物可利用治疗剂。
更新日期:2018-05-22
中文翻译:
作为单胺氧化酶抑制剂的新型吡咯衍生物的设计、合成、体外和计算机评估
为了开发有效的单胺氧化酶 (MAO) 抑制剂,通过选择性还原 1-(1-甲基-1H-吡咯-2-基)-3-[5- (芳基)呋喃-2-基]丙-2-烯-1-酮通过钯催化在乙醇中加氢。通过体外荧光法筛选合成的化合物对 MAO-A 和 MAO-B 的抑制作用。选择性指数 (SI) 值以每种化合物的 IC50 (MAO-A)/IC50 (MAO-B) 的比率给出。3-(5-(4-Chlorophenyl)furan-2-yl)-1-(1-methyl-1H-pyrrol-2-yl)propan-1-one (6) 被鉴定为最具选择性的 MAO-A 抑制剂在该系列中,IC50 值为 0.162 µM,SI 值为 0.002。还进行了动力学研究以评估化合物 6 对 MAO-A 抑制的性质。 根据 Lineweaver-Burk 图,发现化合物 6 是一种竞争性 MAO-A 抑制剂,化合物 6 的 Ki 值确定为 0.1221 μM。使用人 MAO-A 晶体结构(PDB ID:2Z5Y)对化合物 6 和氯吉林进行对接研究。对接结果表明,化合物6在酶的活性中心腔中表现出与氯吉林相似的相互作用。Molinspiration 软件用于确定所有化合物的理化参数,以评估它们是否符合 Lipinski 的五法则。化合物 6 没有违反 Lipinski 规则,使其成为潜在的口服生物可利用治疗剂。对接结果表明,化合物6在酶的活性中心腔中表现出与氯吉林相似的相互作用。Molinspiration 软件用于确定所有化合物的理化参数,以评估它们是否符合 Lipinski 的五法则。化合物 6 没有违反 Lipinski 规则,使其成为潜在的口服生物可利用治疗剂。对接结果表明,化合物6在酶的活性中心腔中表现出与氯吉林相似的相互作用。Molinspiration 软件用于确定所有化合物的理化参数,以评估它们是否符合 Lipinski 的五法则。化合物 6 没有违反 Lipinski 规则,使其成为潜在的口服生物可利用治疗剂。














































 京公网安备 11010802027423号
京公网安备 11010802027423号