当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Adsorption and Diffusion of Lithium on Monolayer Transition Metal Dichalcogenides (MoS2(1–x)Se2x) Alloys
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2015-12-10 00:00:00 , DOI: 10.1021/acs.jpcc.5b09034 Fatih Ersan 1 , Gökhan Gökoğlu 2 , Ethem Aktürk 1, 3
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2015-12-10 00:00:00 , DOI: 10.1021/acs.jpcc.5b09034 Fatih Ersan 1 , Gökhan Gökoğlu 2 , Ethem Aktürk 1, 3
Affiliation
|
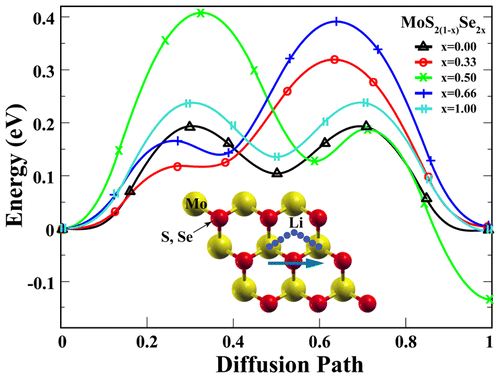
On the basis of first-principles plane-wave calculations, we examine the adsorption and diffusion of lithium on the hexagonal MoS2(1–x)Se2x monolayers with variation of x for 0.00, 0.33, 0.50, 0.66, and 1.00. We find that the lowest energy adsorption positions of Li adatom is at the top site of Mo atom in both MoS2 and MoSe2 monolayers, while Li moves through the Mo—S bond for MoS2(1–x)Se2x. While bare MoS2(1–x)Se2x compounds are nonmagnetic semiconductor and its energy band gap varies with x, they can be metallized by Li adsorption. NEB calculation results show that their energy barriers make them suitable for using in electrode materials. The lithium adsorption energy is sensitive to the external strain, when we elongate the lattice constants, the adsorption energy decreases quickly. We also examine the penetration energy barrier for single lithium atom to pass through the MoS2(1–x)Se2x monolayers, this barrier is decreasing from ∼2.5 eV to ∼1.3 eV with increasing selenium concentration.
中文翻译:

锂在单层过渡金属双硫属元素化物(MoS 2(1– x) Se 2 x)合金上的吸附和扩散
上第一原理平面波计算的基础上,我们检查在六角形的MoS吸附和锂的扩散2(1- X)硒2 X单层用的变异X为0.00,0.33,0.50,0.66,和1.00。我们发现,Li原子的最低能量吸附位置在MoS 2和MoSe 2单层中的Mo原子的顶部,而Li穿过MoS 2(1- x) Se 2 x的Mo-S键移动。MoS 2(1– x) Se 2 x裸化合物是非磁性半导体,其能带隙随x,它们可以通过Li吸附而金属化。NEB计算结果表明,它们的能垒使它们适合用于电极材料。锂的吸附能对外部应变敏感,当我们延长晶格常数时,吸附能迅速下降。我们还研究了单个锂原子穿过MoS 2(1- x) Se 2 x单层的渗透能垒,该垒能随着硒浓度的增加而从约2.5 eV降至约1.3 eV。
更新日期:2015-12-10
中文翻译:
锂在单层过渡金属双硫属元素化物(MoS 2(1– x) Se 2 x)合金上的吸附和扩散
上第一原理平面波计算的基础上,我们检查在六角形的MoS吸附和锂的扩散2(1- X)硒2 X单层用的变异X为0.00,0.33,0.50,0.66,和1.00。我们发现,Li原子的最低能量吸附位置在MoS 2和MoSe 2单层中的Mo原子的顶部,而Li穿过MoS 2(1- x) Se 2 x的Mo-S键移动。MoS 2(1– x) Se 2 x裸化合物是非磁性半导体,其能带隙随x,它们可以通过Li吸附而金属化。NEB计算结果表明,它们的能垒使它们适合用于电极材料。锂的吸附能对外部应变敏感,当我们延长晶格常数时,吸附能迅速下降。我们还研究了单个锂原子穿过MoS 2(1- x) Se 2 x单层的渗透能垒,该垒能随着硒浓度的增加而从约2.5 eV降至约1.3 eV。
































 京公网安备 11010802027423号
京公网安备 11010802027423号