当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Computational Studies of Aromatic and Photophysical Properties of Expanded Porphyrins
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-05-09 00:00:00 , DOI: 10.1021/acs.jpca.8b02311 Rashid R. Valiev 1, 2 , Isaac Benkyi 1 , Yuri V. Konyshev 2 , Heike Fliegl 3 , Dage Sundholm 1, 4
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-05-09 00:00:00 , DOI: 10.1021/acs.jpca.8b02311 Rashid R. Valiev 1, 2 , Isaac Benkyi 1 , Yuri V. Konyshev 2 , Heike Fliegl 3 , Dage Sundholm 1, 4
Affiliation
|
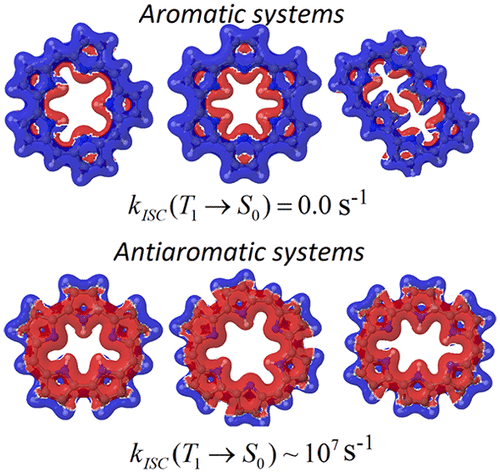
Magnetically induced current densities and ring-current pathways have been calculated at density functional theory (DFT) and second-order Møller–Plesset perturbation theory (MP2) levels of theory for a set of expanded porphyrins consisting of five or six pyrrolic rings. The studied molecules are sapphyrin, cyclo[6]pyrrole, rubyrin, orangarin, rosarin, and amethyrin. Different functionals have been employed to assess the functional dependence of the ring-current strength susceptibility. Vertical singlet and triplet excitation energies have been calculated at the second-order approximate coupled cluster (CC2), expanded multiconfigurational quasi-degenerate perturbation theory (XMC-DPT2), and time-dependent density functional theory levels. The lowest electronic transition of the antiaromatic molecules was found to be pure magnetic transitions providing an explanation for the large paratropic contribution to the total current density. Rate constants for different nonradiative deactivation channels of the lowest excited states have been calculated yielding lifetimes and quantum yields of the lowest excited singlet and triplet states. The calculations show that the spin–orbit interaction between the lowest singlet (S0) and triplet (T1) states of the antiaromatic molecules is strong, whereas for the aromatic molecule the spin–orbit coupling vanishes. The experimentally detected fluorescence from S2 to S0 of amethyrin has been explained. The study shows that there are correlations between the aromatic character and optical properties of the investigated expanded porphyrins.
中文翻译:

膨胀卟啉的芳香和光物理性质的计算研究
对于一组由五个或六个吡咯环组成的扩展卟啉,已根据密度泛函理论(DFT)和二阶Møller-Plesset微扰理论(MP2)的理论水平计算了磁感应电流密度和环电流路径。所研究的分子是沙弗林,环[6]吡咯,红宝石素,猩红素,罗沙林和甲基菊酯。已经采用了不同的功能来评估环电流强度敏感性的功能依赖性。垂直单重态和三重态激发能已在二阶近似耦合簇(CC2),扩展的多构型拟简简扰动理论(XMC-DPT2)和时间依赖的密度泛函理论水平上进行了计算。发现抗芳族分子的最低电子跃迁是纯磁跃迁,这为对总电流密度的大副边形贡献做出了解释。已计算出最低激发态的不同非辐射失活通道的速率常数,从而得出了最低激发单重态和三重态的寿命和量子产率。计算表明,最低单重态之间的自旋-轨道相互作用(抗芳族分子的S 0)和三重态(T 1)状态很强,而对于芳族分子,自旋-轨道耦合消失。已经解释了从实验上检测到的从甲基银的S 2到S 0的荧光。研究表明,所研究的膨胀卟啉的芳香特性与光学性质之间存在相关性。
更新日期:2018-05-09
中文翻译:
膨胀卟啉的芳香和光物理性质的计算研究
对于一组由五个或六个吡咯环组成的扩展卟啉,已根据密度泛函理论(DFT)和二阶Møller-Plesset微扰理论(MP2)的理论水平计算了磁感应电流密度和环电流路径。所研究的分子是沙弗林,环[6]吡咯,红宝石素,猩红素,罗沙林和甲基菊酯。已经采用了不同的功能来评估环电流强度敏感性的功能依赖性。垂直单重态和三重态激发能已在二阶近似耦合簇(CC2),扩展的多构型拟简简扰动理论(XMC-DPT2)和时间依赖的密度泛函理论水平上进行了计算。发现抗芳族分子的最低电子跃迁是纯磁跃迁,这为对总电流密度的大副边形贡献做出了解释。已计算出最低激发态的不同非辐射失活通道的速率常数,从而得出了最低激发单重态和三重态的寿命和量子产率。计算表明,最低单重态之间的自旋-轨道相互作用(抗芳族分子的S 0)和三重态(T 1)状态很强,而对于芳族分子,自旋-轨道耦合消失。已经解释了从实验上检测到的从甲基银的S 2到S 0的荧光。研究表明,所研究的膨胀卟啉的芳香特性与光学性质之间存在相关性。
































 京公网安备 11010802027423号
京公网安备 11010802027423号