当前位置:
X-MOL 学术
›
ACS Catal.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Computational Mechanism Study of Catalyst-Dependent Competitive 1,2-C→C, −O→C, and −N→C Migrations from β-Methylene-β-silyloxy-β-amido-α-diazoacetate: Insight into the Origins of Chemoselectivity
ACS Catalysis ( IF 11.3 ) Pub Date : 2015-12-04 00:00:00 , DOI: 10.1021/acscatal.5b02103 Xin Yang 1 , Yongsheng Yang 1 , Ying Xue 1
ACS Catalysis ( IF 11.3 ) Pub Date : 2015-12-04 00:00:00 , DOI: 10.1021/acscatal.5b02103 Xin Yang 1 , Yongsheng Yang 1 , Ying Xue 1
Affiliation
|
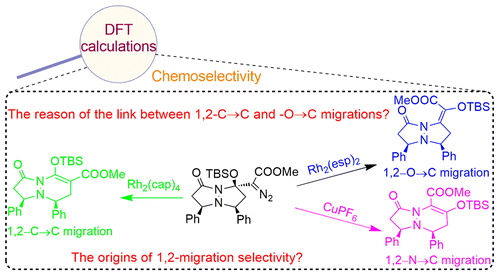
Doyle et al. [ J. Am. Chem. Soc. 2013, 135, 1244−1247] recently reported an efficient catalyst-controlled chemoselectivity of competitive 1,2-C→C, −O→C, and −N→C migrations from β-methylene-β-silyloxy-β-amido-α-diazoacetates using dirhodium or copper catalysts. With the aid of density functional theory calculations, the present study systematically probed the mechanism of the aforementioned reactions and the origins of the catalyst-controlled chemoselectivity. Similar to the method reported in the literature, simplified catalyst models Rh2(O2CH)4 and Rh2(N-methylformamide)4 have been used in our initial calculations. However, using the Rh2(O2CH)4 model could not describe the energies of all possible pathways, and high selectivity of three competitive migrations could not be achieved. In order to appropriately describe this 1,2-migration system, real catalyst models Rh2(cap)4, Rh2(esp)2, and CuPF6 have been employed. It was found that the steric and electronic effects of ligands significantly influence the free energy barrier, which ultimately changes the chemoselectivity. In the CuPF6 system, the electronic effects, coupled with the steric factor, give a qualitative explanation for the exclusive chemoselectivity of 1,2-N→C migration over 1,2-C→C or −O→C migration. On the other hand, the bulky ligands of dirhodium catalysts result in the significant steric hindrance around the dirhodium centers and withdrawal of the empty space around the bulky −OTBS group. By analyzing the divergence of three different migration transition states using the distortion/interaction and natural bond orbital analyses, it was found that the 1,2-N→C migration will suffer from a high free energy barrier because of the steric repulsion between the carbonyl group and the carbonyl oxygen of the pyrazolidinone ring. For 1,2-C→C and −O→C migrations, changing the ligands of dirhodium catalysts can change the electronic properties of carbenes, and that is the reason for controlling the major product by changing the dirhodium catalysts. The mechanistic proposal is supported by the calculated chemoselectivities, which are in good agreement with the experimental results.
中文翻译:

β-亚甲基-β-甲硅烷氧基-β-酰胺基-α-二重氮乙酸酯的催化剂依赖性竞争性1,2-C→C,-O→C和-N→C迁移的计算机理研究:对化学选择性的认识
Doyle等。[ J. Am。化学 Soc。 2013,135,1244年至1247年]最近报道的有竞争力的1,2--C→C,-O→C,和-N→C从迁移的有效催化剂控制的化学选择性β亚甲基- β-甲硅烷氧基-β-酰氨基使用二氢吡啶鎓或铜催化剂的α-重氮乙酸盐。借助密度泛函理论计算,本研究系统地探讨了上述反应的机理以及催化剂控制的化学选择性的起源。与文献报道的方法类似,简化的催化剂模型Rh 2(O 2 CH)4和Rh 2(N-甲基甲酰胺)4已用于我们的初始计算中。但是,使用Rh 2(O 2 CH)4模型无法描述所有可能途径的能量,并且无法实现三种竞争性迁移的高选择性。为了适当地描述该1,2-迁移系统,已经使用了真实的催化剂模型Rh 2(cap)4,Rh 2(esp)2和CuPF 6。发现配体的空间和电子效应显着影响自由能垒,其最终改变化学选择性。在CuPF 6中在系统中,电子效应与空间位阻相结合,定性地解释了1,2-N→C迁移在1,2-C→C或-O→C迁移方面的排他性化学选择性。另一方面,dirhodium催化剂的庞大配体会导致在dirhodium中心周围出现明显的空间位阻,并导致庞大的-OTBS基团周围的空白区域消失。通过畸变/相互作用和自然键轨道分析,分析了三种不同迁移过渡态的发散,发现由于羰基之间的空间排斥,1,2-N→C迁移将受到高自由能垒的影响。基和吡唑烷酮环的羰基氧。对于1,2-C→C和-O→C迁移,改变dirhodium催化剂的配体会改变碳烯的电子性质,这就是通过更换铱催化剂来控制主要产品的原因。机理建议由计算的化学选择性支持,与实验结果非常吻合。
更新日期:2015-12-04
中文翻译:
β-亚甲基-β-甲硅烷氧基-β-酰胺基-α-二重氮乙酸酯的催化剂依赖性竞争性1,2-C→C,-O→C和-N→C迁移的计算机理研究:对化学选择性的认识
Doyle等。[ J. Am。化学 Soc。 2013,135,1244年至1247年]最近报道的有竞争力的1,2--C→C,-O→C,和-N→C从迁移的有效催化剂控制的化学选择性β亚甲基- β-甲硅烷氧基-β-酰氨基使用二氢吡啶鎓或铜催化剂的α-重氮乙酸盐。借助密度泛函理论计算,本研究系统地探讨了上述反应的机理以及催化剂控制的化学选择性的起源。与文献报道的方法类似,简化的催化剂模型Rh 2(O 2 CH)4和Rh 2(N-甲基甲酰胺)4已用于我们的初始计算中。但是,使用Rh 2(O 2 CH)4模型无法描述所有可能途径的能量,并且无法实现三种竞争性迁移的高选择性。为了适当地描述该1,2-迁移系统,已经使用了真实的催化剂模型Rh 2(cap)4,Rh 2(esp)2和CuPF 6。发现配体的空间和电子效应显着影响自由能垒,其最终改变化学选择性。在CuPF 6中在系统中,电子效应与空间位阻相结合,定性地解释了1,2-N→C迁移在1,2-C→C或-O→C迁移方面的排他性化学选择性。另一方面,dirhodium催化剂的庞大配体会导致在dirhodium中心周围出现明显的空间位阻,并导致庞大的-OTBS基团周围的空白区域消失。通过畸变/相互作用和自然键轨道分析,分析了三种不同迁移过渡态的发散,发现由于羰基之间的空间排斥,1,2-N→C迁移将受到高自由能垒的影响。基和吡唑烷酮环的羰基氧。对于1,2-C→C和-O→C迁移,改变dirhodium催化剂的配体会改变碳烯的电子性质,这就是通过更换铱催化剂来控制主要产品的原因。机理建议由计算的化学选择性支持,与实验结果非常吻合。

















































 京公网安备 11010802027423号
京公网安备 11010802027423号