当前位置:
X-MOL 学术
›
ChemistrySelect
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Indolocarbazole (IC) Derivatives as Promising p‐type Organic Semiconductors: A First‐Principle Study of Their Anisotropic Charge Mobilities
ChemistrySelect ( IF 1.9 ) Pub Date : 2018-05-02 , DOI: 10.1002/slct.201800285
Smruti R. Sahoo 1 , Sridhar Sahu 1 , Sagar Sharma 2
ChemistrySelect ( IF 1.9 ) Pub Date : 2018-05-02 , DOI: 10.1002/slct.201800285
Smruti R. Sahoo 1 , Sridhar Sahu 1 , Sagar Sharma 2
Affiliation
|
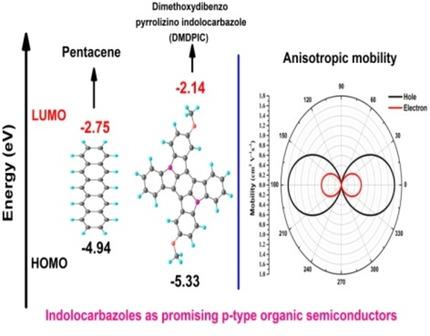
We present a detailed theoretical study of the structural, charge transport, and optical properties of three indolocarbazole derivatives, viz., 11, 12‐Dihydroindolo[2, 3‐a]carbazole (DIC), 5, 10‐dimethyl‐5,10‐dihydrobenzo[a]indolo[2, 3‐c]carbazole (DMDBIC), and 2,11‐dimethoxydibenzo[2, 3:5,6]pyrrolizino[1, 7‐bc]indolo[1, 2,3‐lm]carbazole (DMDPIC). DFT calculations along with previously reported x‐ray crystal structures reveal that the studied compounds are planar and do not undergo significant changes in structure upon oxidation or reduction. The maximum hole anisotropic charge mobilities for DIC was found to be 0.338 cm2 V−1 s−1 at Φ=175.89° and 355.81° and the maximum electron mobility was 0.181 cm2 V−1 s−1 at Φ=85.94° and 265.85°. For DMDBIC, the predicted maximum anisotropic hole, and electron mobility was 0.501 cm2 V−1 s−1 and 0.775 cm2 V−1 s−1, respectively at Φ=56.72° and 236.63°, while for DMDPIC the predicted maximum anisotropic hole and electron mobility as 1.586 cm2 V−1 s−1 and 0.594 cm2 V−1 s−1, respectively at Φ=0° and 180.0°. The calculated mobilities show that these compounds may be suitable candidates as a p‐type organic semiconductor with better stability than acene compounds.
中文翻译:

吲哚并咔唑(IC)衍生物作为有前景的p型有机半导体:各向异性电荷迁移率的第一性原理研究
我们对三种吲哚并咔唑衍生物的结构,电荷传输和光学性质进行了详细的理论研究,即11,11,12-二氢吲哚并[2,3-a]咔唑(DIC),5,10-二甲基-5,10 -二氢苯并[a]吲哚并[2,3-c]咔唑(DMDBIC)和2,11-二甲氧基二苯并[2,3:5,6]吡咯啉并[1,7-bc]吲并[1,2,3-lm ]咔唑(DMDPIC)。DFT计算以及先前报道的X射线晶体结构表明,所研究的化合物是平面的,在氧化或还原后结构不会发生重大变化。发现在Φ = 175.89°和355.81°下,DIC的最大空穴各向异性电荷迁移率是0.338 cm 2 V -1 s -1,最大电子迁移率是0.181 cm 2 V-1小号-1在Φ = 85.94°和265.85°。对于DMDBIC,在Φ = 56.72°和236.63°时,预测的最大各向异性空穴和电子迁移率分别为0.501 cm 2 V -1 s -1和0.775 cm 2 V -1 s -1,而对于DMDPIC,预测的最大各向异性在Φ处的空穴和电子迁移率分别为1.586 cm 2 V -1 s -1和0.594 cm 2 V -1 s -1= 0°和180.0°。计算出的迁移率表明,这些化合物可能是适合作为ap型有机半导体的候选物,其稳定性要比并苯化合物更好。
更新日期:2018-05-02
中文翻译:
吲哚并咔唑(IC)衍生物作为有前景的p型有机半导体:各向异性电荷迁移率的第一性原理研究
我们对三种吲哚并咔唑衍生物的结构,电荷传输和光学性质进行了详细的理论研究,即11,11,12-二氢吲哚并[2,3-a]咔唑(DIC),5,10-二甲基-5,10 -二氢苯并[a]吲哚并[2,3-c]咔唑(DMDBIC)和2,11-二甲氧基二苯并[2,3:5,6]吡咯啉并[1,7-bc]吲并[1,2,3-lm ]咔唑(DMDPIC)。DFT计算以及先前报道的X射线晶体结构表明,所研究的化合物是平面的,在氧化或还原后结构不会发生重大变化。发现在Φ = 175.89°和355.81°下,DIC的最大空穴各向异性电荷迁移率是0.338 cm 2 V -1 s -1,最大电子迁移率是0.181 cm 2 V-1小号-1在Φ = 85.94°和265.85°。对于DMDBIC,在Φ = 56.72°和236.63°时,预测的最大各向异性空穴和电子迁移率分别为0.501 cm 2 V -1 s -1和0.775 cm 2 V -1 s -1,而对于DMDPIC,预测的最大各向异性在Φ处的空穴和电子迁移率分别为1.586 cm 2 V -1 s -1和0.594 cm 2 V -1 s -1= 0°和180.0°。计算出的迁移率表明,这些化合物可能是适合作为ap型有机半导体的候选物,其稳定性要比并苯化合物更好。
































 京公网安备 11010802027423号
京公网安备 11010802027423号