当前位置:
X-MOL 学术
›
J. Phys. Chem. Lett.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
IR Spectra of (HCOOH)2 and (DCOOH)2: Experiment, VSCF/VCI, and Ab Initio Molecular Dynamics Calculations Using Full-Dimensional Potential and Dipole Moment Surfaces
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2018-04-30 00:00:00 , DOI: 10.1021/acs.jpclett.8b00447 Chen Qu 1 , Joel M. Bowman 1
The Journal of Physical Chemistry Letters ( IF 4.8 ) Pub Date : 2018-04-30 00:00:00 , DOI: 10.1021/acs.jpclett.8b00447 Chen Qu 1 , Joel M. Bowman 1
Affiliation
|
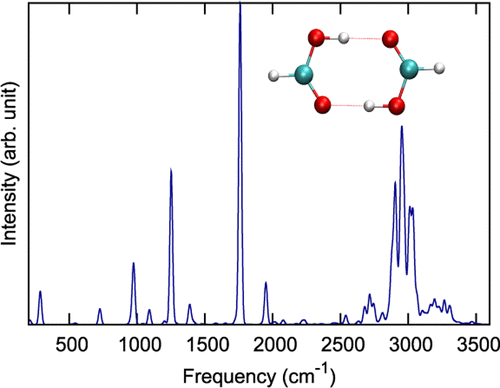
We report quantum VSCF/VCI and ab initio molecular dynamics (AIMD) calculations of the IR spectra of (HCOOH)2 and (DCOOH)2, using full-dimensional, ab initio potential energy and dipole moment surfaces (PES and DMS). These surfaces are fits, using permutationally invariant polynomials, to 13 475 ab initio CCSD(T)-F12a electronic energies and MP2 dipole moments. Here “AIMD” means using these ab initio potential and dipole moment surfaces in the MD calculations. The VSCF/VCI calculations use all (24) normal modes for coupling, with a four-mode representation of the potential. The quantum spectra align well with jet-cooled and room-temperature experimental spectra over the spectral range 600–3600 cm–1. Analyses of the complex O–H and C–H stretch bands are made based on the mixing of the VSCF/VCI basis functions. The comparisons of the AIMD IR spectra with both experimental and VSCF/VCI ones provide tests of the accuracy of the AIMD approach. These indicate good accuracy for simple bands but not for the complex O–H stretch band, which is upshifted from experimental and VSCF/VCI bands by roughly 300 cm–1. In addition to testing the AIMD approach, the PES, DMS, and VSCF/VCI calculations for formic acid dimer provide opportunities for testing other methods to represent high-dimensional data and other methods that perform postharmonic vibrational calculations.
中文翻译:

(HCOOH)2和(DCOOH)2的红外光谱:实验,VSCF / VCI和使用全尺寸势和偶极矩表面进行的从头算分子动力学计算
我们使用全尺寸,从头算势能和偶极矩表面(PES和DMS)报告了(HCOOH)2和(DCOOH)2的IR光谱的量子VSCF / VCI和从头算分子动力学(AIMD)计算。这些表面使用置换不变多项式拟合从头开始的13 475 CCSD(T)-F12a电子能量和MP2偶极矩。在此,“ AIMD”是指在MD计算中使用这些从头算势和偶极矩表面。VSCF / VCI计算使用所有(24)正常模式进行耦合,并以四模式表示电势。在600–3600 cm –1的光谱范围内,量子光谱与射流冷却和室温的实验光谱完全吻合。基于VSCF / VCI基函数的混合,对复杂的O–H和C–H伸缩带进行了分析。AIMD红外光谱与实验和VSCF / VCI的比较提供了AIMD方法准确性的测试。这些表明简单频段的精度很高,但复杂的OH拉伸频段则没有,后者从实验频段和VSCF / VCI频段上移了大约300 cm –1。除了测试AIMD方法外,甲酸二聚体的PES,DMS和VSCF / VCI计算还提供了测试代表高维数据的其他方法以及执行谐波后振动计算的其他方法的机会。
更新日期:2018-04-30
中文翻译:
(HCOOH)2和(DCOOH)2的红外光谱:实验,VSCF / VCI和使用全尺寸势和偶极矩表面进行的从头算分子动力学计算
我们使用全尺寸,从头算势能和偶极矩表面(PES和DMS)报告了(HCOOH)2和(DCOOH)2的IR光谱的量子VSCF / VCI和从头算分子动力学(AIMD)计算。这些表面使用置换不变多项式拟合从头开始的13 475 CCSD(T)-F12a电子能量和MP2偶极矩。在此,“ AIMD”是指在MD计算中使用这些从头算势和偶极矩表面。VSCF / VCI计算使用所有(24)正常模式进行耦合,并以四模式表示电势。在600–3600 cm –1的光谱范围内,量子光谱与射流冷却和室温的实验光谱完全吻合。基于VSCF / VCI基函数的混合,对复杂的O–H和C–H伸缩带进行了分析。AIMD红外光谱与实验和VSCF / VCI的比较提供了AIMD方法准确性的测试。这些表明简单频段的精度很高,但复杂的OH拉伸频段则没有,后者从实验频段和VSCF / VCI频段上移了大约300 cm –1。除了测试AIMD方法外,甲酸二聚体的PES,DMS和VSCF / VCI计算还提供了测试代表高维数据的其他方法以及执行谐波后振动计算的其他方法的机会。





























 京公网安备 11010802027423号
京公网安备 11010802027423号