当前位置:
X-MOL 学术
›
J. Phys. Chem. A
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Dissociation Energy of the H2O···HF Dimer
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-04-18 00:00:00 , DOI: 10.1021/acs.jpca.8b03397 Thomas More Sexton 1 , J. Coleman Howard 2 , Gregory S. Tschumper 1
The Journal of Physical Chemistry A ( IF 2.7 ) Pub Date : 2018-04-18 00:00:00 , DOI: 10.1021/acs.jpca.8b03397 Thomas More Sexton 1 , J. Coleman Howard 2 , Gregory S. Tschumper 1
Affiliation
|
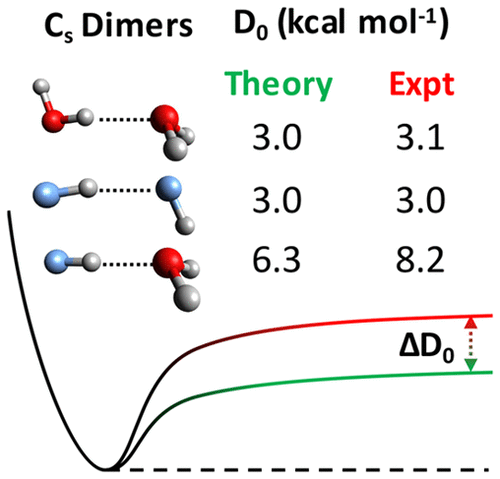
Even though (H2O)2 and (HF)2 are arguably the most thoroughly characterized prototypes for hydrogen bonding, their heterogeneous analogue H2O···HF has received relatively little attention. Here we report that the experimental dissociation energy (D0) of this important paradigm for heterogeneous hydrogen bonding is too large by 2 kcal mol–1 or 30% relative to our computed value of 6.3 kcal mol–1. For reference, computational procedures similar to those employed here to compute D0 (large basis set CCSD(T) computations with anharmonic corrections from second-order vibrational perturbation theory) provide results within 0.1 kcal mol–1 of the experimental values for (H2O)2 and (HF)2. Near the CCSD(T) complete basis set limit, the electronic dissociation energy for H2O···HF is ∼4 kcal mol–1 larger than those for (H2O)2 and (HF)2 (∼9 kcal mol–1 for the heterogeneous dimer vs ∼5 kcal mol–1 for the homogeneous dimers). Results reported here from symmetry-adapted perturbation theory computations suggest that this large difference is primarily due to the induction contribution to the interaction energy.
中文翻译:

H 2 O···HF二聚体的离解能
尽管(H 2 O)2和(HF)2可以说是最充分表征氢键的原型,但它们的异质类似物H 2 O··HF却很少受到关注。在这里,我们报道了这个重要的非均质氢键范式的实验解离能(D 0)相对于我们的6.3 kcal mol -1的计算值太大了2 kcal mol –1或30%。作为参考,与此处用于计算D 0的计算过程相似的计算过程(使用二阶振动摄动理论进行非谐校正的大基数CCSD(T)计算)可提供(H 2 O)2和(HF)2实验值在0.1 kcal mol –1范围内的结果。附近的CCSD(T)完成基组限制,对于H中的电子离解能2 ö···HF为约为4千卡摩尔-1比那些(H较大2 O)2和(HF)2(〜9千卡摩尔–1对于异质二聚体与〜5 kcal mol –1对于均匀的二聚体)。对称适应的扰动理论计算在此报告的结果表明,这种较大的差异主要归因于感应对相互作用能的贡献。
更新日期:2018-04-18
中文翻译:
H 2 O···HF二聚体的离解能
尽管(H 2 O)2和(HF)2可以说是最充分表征氢键的原型,但它们的异质类似物H 2 O··HF却很少受到关注。在这里,我们报道了这个重要的非均质氢键范式的实验解离能(D 0)相对于我们的6.3 kcal mol -1的计算值太大了2 kcal mol –1或30%。作为参考,与此处用于计算D 0的计算过程相似的计算过程(使用二阶振动摄动理论进行非谐校正的大基数CCSD(T)计算)可提供(H 2 O)2和(HF)2实验值在0.1 kcal mol –1范围内的结果。附近的CCSD(T)完成基组限制,对于H中的电子离解能2 ö···HF为约为4千卡摩尔-1比那些(H较大2 O)2和(HF)2(〜9千卡摩尔–1对于异质二聚体与〜5 kcal mol –1对于均匀的二聚体)。对称适应的扰动理论计算在此报告的结果表明,这种较大的差异主要归因于感应对相互作用能的贡献。






























 京公网安备 11010802027423号
京公网安备 11010802027423号