当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
AMOEBA Polarizable Force Field Parameters of the Heme Cofactor in Its Ferrous and Ferric Forms
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-04-09 00:00:00 , DOI: 10.1021/acs.jctc.7b01128 Xiaojing Wu 1 , Carine Clavaguera 1 , Louis Lagardère 2, 3 , Jean-Philip Piquemal 4, 5, 6 , Aurélien de la Lande 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-04-09 00:00:00 , DOI: 10.1021/acs.jctc.7b01128 Xiaojing Wu 1 , Carine Clavaguera 1 , Louis Lagardère 2, 3 , Jean-Philip Piquemal 4, 5, 6 , Aurélien de la Lande 1
Affiliation
|
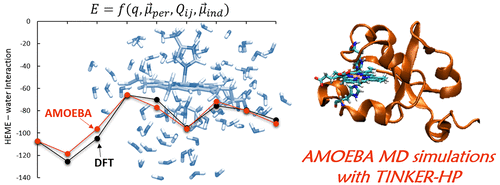
We report the first parameters of the heme redox cofactors for the polarizable AMOEBA force field in both the ferric and ferrous forms. We consider two types of complexes, one with two histidine side chains as axial ligands and one with a histidine and a methionine side chain as ligands. We have derived permanent multipoles from second-order Møller–Plesset perturbation theory (MP2). The sets of parameters have been validated in a first step by comparison of AMOEBA interaction energies of heme and a collection of biologically relevant molecules with MP2 and Density Functional Theory (DFT) calculations. In a second validation step, we consider interaction energies with large aggregates comprising around 80 H2O molecules. These calculations are repeated for 30 structures extracted from semiempirical PM7 DM simulations. Very encouraging agreement is found between DFT and the AMOEBA force field, which results from an accurate treatment of electrostatic interactions. We finally report long (10 ns) MD simulations of cytochromes in two redox states with AMOEBA testing both the 2003 and 2014 AMOEBA water models. These simulations have been carried out with the TINKER-HP (High Performance) program. In conclusion, owing to their ubiquity in biology, we think the present work opens a wide array of applications of the polarizable AMOEBA force field on hemeproteins.
中文翻译:

亚铁和铁形式血红素辅因子的AMOEBA可极化力场参数
我们报告了铁和亚铁形式的可极化AMOEBA力场的血红素氧化还原辅助因子的第一个参数。我们考虑两种类型的复合物,一种以两个组氨酸侧链为轴向配体,另一种以组氨酸和甲硫氨酸侧链为配体。我们从二阶Møller–Plesset摄动理论(MP2)中得出了永久多极子。通过比较血红素的AMOEBA相互作用能和具有MP2和密度泛函理论(DFT)计算的生物学上相关的分子的集合,已在第一步中验证了参数集。在第二个验证步骤中,我们考虑与包含约80 H 2的大聚集体的相互作用能O分子。对于从半经验PM7 DM模拟中提取的30个结构重复了这些计算。在DFT和AMOEBA力场之间发现了非常令人鼓舞的协议,这是对静电相互作用的精确处理所导致的。我们最终报告了两个氧化还原状态下细胞色素的长时间(10 ns)MD模拟,其中AMOEBA测试了2003年和2014年AMOEBA水模型。这些模拟是使用TINKER-HP(高性能)程序进行的。总之,由于它们在生物学中的普遍性,我们认为当前的研究为极化蛋白AMOEBA力场在血红蛋白上的广泛应用开辟了道路。
更新日期:2018-04-09
中文翻译:
亚铁和铁形式血红素辅因子的AMOEBA可极化力场参数
我们报告了铁和亚铁形式的可极化AMOEBA力场的血红素氧化还原辅助因子的第一个参数。我们考虑两种类型的复合物,一种以两个组氨酸侧链为轴向配体,另一种以组氨酸和甲硫氨酸侧链为配体。我们从二阶Møller–Plesset摄动理论(MP2)中得出了永久多极子。通过比较血红素的AMOEBA相互作用能和具有MP2和密度泛函理论(DFT)计算的生物学上相关的分子的集合,已在第一步中验证了参数集。在第二个验证步骤中,我们考虑与包含约80 H 2的大聚集体的相互作用能O分子。对于从半经验PM7 DM模拟中提取的30个结构重复了这些计算。在DFT和AMOEBA力场之间发现了非常令人鼓舞的协议,这是对静电相互作用的精确处理所导致的。我们最终报告了两个氧化还原状态下细胞色素的长时间(10 ns)MD模拟,其中AMOEBA测试了2003年和2014年AMOEBA水模型。这些模拟是使用TINKER-HP(高性能)程序进行的。总之,由于它们在生物学中的普遍性,我们认为当前的研究为极化蛋白AMOEBA力场在血红蛋白上的广泛应用开辟了道路。






























 京公网安备 11010802027423号
京公网安备 11010802027423号