当前位置:
X-MOL 学术
›
Inorg. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Electron-Transfer-Enhanced Cation–Cation Interactions in Homo- and Heterobimetallic Actinide Complexes: A Relativistic Density Functional Theory Study
Inorganic Chemistry ( IF 4.3 ) Pub Date : 2018-03-21 00:00:00 , DOI: 10.1021/acs.inorgchem.8b00051 Ming Zheng 1 , Fang-Yuan Chen 1 , Jia-Nan Tian 1 , Qing-Jiang Pan 1
Inorganic Chemistry ( IF 4.3 ) Pub Date : 2018-03-21 00:00:00 , DOI: 10.1021/acs.inorgchem.8b00051 Ming Zheng 1 , Fang-Yuan Chen 1 , Jia-Nan Tian 1 , Qing-Jiang Pan 1
Affiliation
|
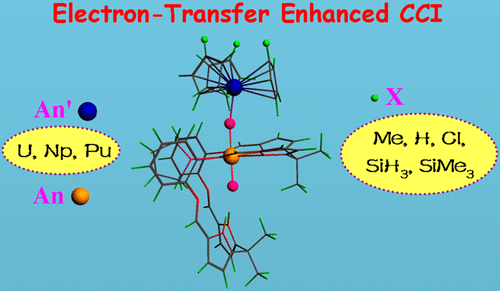
To provide deep insight into cation–cation interactions (CCIs) involving hexavalent actinyl species that are major components in spent nuclear fuel and pose important implications for the effective removal of radiotoxic pollutants in the environment, a series of homo- and heterobimetallic actinide complexes supported by cyclopentadienyl (Cp) and polypyrrolic macrocycle (H4L) ligands were systematically investigated using relativistic density functional theory. The metal sort in both parts of (THF)(H2L)(OAnVIO) and (An′)IIICp3 from U to Np to Pu, as well as the substituent bonding to Cp from electron-donating Me to H to electron-withdrawing Cl, SiH3, and SiMe3, was changed. Over 0.70 electrons are unraveled to transfer from the electron-rich UIII to the electron-deficient AnVI of the actinyl moiety, leading to a more stable AnV–UIV isomer; in contrast, uranylneptunium and uranylplutonium complexes behave as electron-resonance structures between VI–III and V–IV. These were further corroborated by geometrical and electronic structures. The energies of CCIs (i.e., Oexo–An′ bonds) were calculated to be −19.6 to −41.2 kcal/mol, affording those of OUO–Np (−23.9 kcal/mol) and OUO–Pu (−19.6 kcal/mol) with less electron transfer (ET) right at the low limit. Topological analyses of the electron density at the Oexo–An′ bond critical points demonstrate that the CCIs are ET or dative bonds in nature. A positive correlation has been built between the CCIs’ strength and corresponding ET amount. It is concluded that the CCIs of Oexo–An′ are driven by the electrostatic attraction between the actinyl oxo atom (negative) and the actinide ion (positive) and enhanced by their ET. Finally, experimental syntheses of (THF)(H2L)(OUVIO)(An′)IIICp3 (An′ = U and Np) were well reproduced by thermodynamic calculations that yielded negative free energies in a tetrahydrofuran solution but a positive one for their uranylplutonium analogue, which was synthetically inaccessible. So, our thermodynamics would provide implications for the synthetic possibility of other theoretically designed bimetallic actinide complexes.
中文翻译:

均相和异双金属Act系化合物中电子转移增强的阳离子间相互作用:相对论密度泛函理论研究
为了深入了解涉及六价act基物种的阳离子-阳离子相互作用(CCI),六价act基物种是乏核燃料的主要成分,对有效去除环境中的放射性有毒污染物具有重要意义,一系列的均一和异双金属act系元素络合物得到了支持相对论密度泛函理论系统地研究了环戊二烯基(Cp)和聚吡咯大环(H 4 L)配体。从(U)到Np到Pu的(THF)(H 2 L)(OAn VI O)和(An')III Cp 3的金属部分以及从给电子的Me到H的Cp的取代基键合吸电子的Cl,SiH 3和SiMe 3, 被改变了。超过0.70的电子被解散,从富电子的U III转移到act基部分的缺电子的An VI,从而导致更稳定的An V –U IV异构体。相反,铀ny和铀lp络合物表现为VI–III和V–IV之间的电子共振结构。几何和电子结构进一步证实了这些。CCI的能量(即O exo –An'键)经计算为-19.6至-41.2 kcal / mol,提供了OUO–Np(−23.9 kcal / mol)和OUO–Pu(-19.6 kcal / mol)的能量)的电子传输量(ET)较低,恰好在下限。O exo处电子密度的拓扑分析–An'键的临界点表明,CCI本质上是ET或dative键。CCI的强度与相应的ET量之间建立了正相关。可以得出结论,O exo –An'的CCI由by基氧代原子(负)和the系离子(正)之间的静电吸引驱动,并通过其ET增强。最后,实验合成了(THF)(H 2 L)(OU VI O)(An')III Cp 3(An'= U和Np)通过热力学计算得到了很好的再现,在四氢呋喃溶液中产生了负的自由能,而对于铀ny类似物却产生了正的自由能,这在合成上是无法获得的。因此,我们的热力学将为其他理论上设计的双金属act系配合物的合成可能性提供启示。
更新日期:2018-03-21
中文翻译:
均相和异双金属Act系化合物中电子转移增强的阳离子间相互作用:相对论密度泛函理论研究
为了深入了解涉及六价act基物种的阳离子-阳离子相互作用(CCI),六价act基物种是乏核燃料的主要成分,对有效去除环境中的放射性有毒污染物具有重要意义,一系列的均一和异双金属act系元素络合物得到了支持相对论密度泛函理论系统地研究了环戊二烯基(Cp)和聚吡咯大环(H 4 L)配体。从(U)到Np到Pu的(THF)(H 2 L)(OAn VI O)和(An')III Cp 3的金属部分以及从给电子的Me到H的Cp的取代基键合吸电子的Cl,SiH 3和SiMe 3, 被改变了。超过0.70的电子被解散,从富电子的U III转移到act基部分的缺电子的An VI,从而导致更稳定的An V –U IV异构体。相反,铀ny和铀lp络合物表现为VI–III和V–IV之间的电子共振结构。几何和电子结构进一步证实了这些。CCI的能量(即O exo –An'键)经计算为-19.6至-41.2 kcal / mol,提供了OUO–Np(−23.9 kcal / mol)和OUO–Pu(-19.6 kcal / mol)的能量)的电子传输量(ET)较低,恰好在下限。O exo处电子密度的拓扑分析–An'键的临界点表明,CCI本质上是ET或dative键。CCI的强度与相应的ET量之间建立了正相关。可以得出结论,O exo –An'的CCI由by基氧代原子(负)和the系离子(正)之间的静电吸引驱动,并通过其ET增强。最后,实验合成了(THF)(H 2 L)(OU VI O)(An')III Cp 3(An'= U和Np)通过热力学计算得到了很好的再现,在四氢呋喃溶液中产生了负的自由能,而对于铀ny类似物却产生了正的自由能,这在合成上是无法获得的。因此,我们的热力学将为其他理论上设计的双金属act系配合物的合成可能性提供启示。





























 京公网安备 11010802027423号
京公网安备 11010802027423号