当前位置:
X-MOL 学术
›
WIREs Comput. Mol. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Information‐theoretic approach in density functional theory and its recent applications to chemical problems
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 16.8 ) Pub Date : 2019-12-30 , DOI: 10.1002/wcms.1461 Chunying Rong 1, 2 , Bin Wang 2 , Dongbo Zhao 3 , Shubin Liu 4
Wiley Interdisciplinary Reviews: Computational Molecular Science ( IF 16.8 ) Pub Date : 2019-12-30 , DOI: 10.1002/wcms.1461 Chunying Rong 1, 2 , Bin Wang 2 , Dongbo Zhao 3 , Shubin Liu 4
Affiliation
|
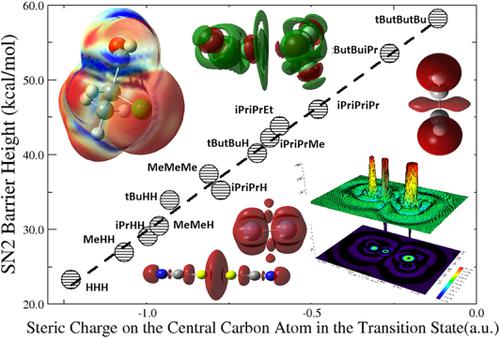
Using simple functionals of the electron density to appreciate and quantify molecular structure and chemical reactivity properties is a recent endeavor in density functional theory (DFT) toward the development of a new chemical reactivity theory. According to the first Hohenberg–Kohn theorem in DFT, the electron density alone should be able to determine any property in the ground state. Exchange and correlation energies are such properties, so are molecular structure and chemical reactivity, and hence they all should accurately be determined by electron density functionals. Quantities such as Shannon entropy, Fisher information, Ghosh–Berkowitz–Parr entropy, information gain, Onicescu information energy, etc., from the information‐theoretic approach are simple electron density functionals, whose analytical forms are exactly known. In this article, we demonstrate their usefulness and validity to quantify regioselectivity, stereoselectivity, and other structure and reactivity properties. We will outline the current understanding of its theoretical framework at first, and then highlight recent applications to chemical problems including isomeric and conformational stability, electrophilicity and nucleophilicity, strong covalent and weak noncovalent interactions, acidity and basicity, aromaticity and antiaromaticity, and numerous other properties. The effort of employing electron density functionals to quantify chemical concepts should open up a new door for us to ultimately develop a chemical reactivity theory using the DFT language.
中文翻译:

密度泛函理论中的信息理论方法及其在化学问题中的最新应用
使用电子密度的简单函数来欣赏和量化分子结构和化学反应性是密度泛函理论(DFT)朝着新化学反应论的发展的最新努力。根据DFT中的第一个Hohenberg-Kohn定理,仅电子密度就能确定基态的任何性质。交换能和相关能都是这种性质,分子结构和化学反应性也是如此,因此它们都应由电子密度函数精确地确定。从信息理论方法来看,诸如香农熵,费舍尔信息,戈什-伯科维茨-帕尔熵,信息增益,奥尼切斯库信息能量等的数量是简单的电子密度泛函,其解析形式是众所周知的。在本文中,我们证明了它们在量化区域选择性,立体选择性以及其他结构和反应性方面的有用性和有效性。我们将首先概述对其理论框架的当前理解,然后重点介绍化学问题的最新应用,包括异构体和构象稳定性,亲电性和亲核性,强共价和弱非共价相互作用,酸度和碱性,芳香性和抗芳香性以及许多其他特性。利用电子密度泛函来量化化学概念的努力应该为我们最终开发使用DFT语言的化学反应性理论打开一扇新门。我们将首先概述对其理论框架的当前理解,然后重点介绍化学问题的最新应用,包括异构体和构象稳定性,亲电性和亲核性,强共价和弱非共价相互作用,酸度和碱性,芳香性和抗芳香性以及许多其他特性。利用电子密度泛函来量化化学概念的努力应该为我们最终开发使用DFT语言的化学反应性理论打开一扇新门。我们将首先概述对其理论框架的当前理解,然后重点介绍化学问题的最新应用,包括异构体和构象稳定性,亲电性和亲核性,强共价和弱非共价相互作用,酸度和碱性,芳香性和抗芳香性以及许多其他特性。利用电子密度泛函来量化化学概念的努力应该为我们最终开发使用DFT语言的化学反应性理论打开一扇新门。芳香性和抗芳香性,以及许多其他特性。利用电子密度泛函来量化化学概念的努力应该为我们最终使用DFT语言发展化学反应性理论打开一扇新门。芳香性和抗芳香性,以及许多其他特性。利用电子密度泛函来量化化学概念的努力应该为我们最终开发使用DFT语言的化学反应性理论打开一扇新门。
更新日期:2019-12-30
中文翻译:
密度泛函理论中的信息理论方法及其在化学问题中的最新应用
使用电子密度的简单函数来欣赏和量化分子结构和化学反应性是密度泛函理论(DFT)朝着新化学反应论的发展的最新努力。根据DFT中的第一个Hohenberg-Kohn定理,仅电子密度就能确定基态的任何性质。交换能和相关能都是这种性质,分子结构和化学反应性也是如此,因此它们都应由电子密度函数精确地确定。从信息理论方法来看,诸如香农熵,费舍尔信息,戈什-伯科维茨-帕尔熵,信息增益,奥尼切斯库信息能量等的数量是简单的电子密度泛函,其解析形式是众所周知的。在本文中,我们证明了它们在量化区域选择性,立体选择性以及其他结构和反应性方面的有用性和有效性。我们将首先概述对其理论框架的当前理解,然后重点介绍化学问题的最新应用,包括异构体和构象稳定性,亲电性和亲核性,强共价和弱非共价相互作用,酸度和碱性,芳香性和抗芳香性以及许多其他特性。利用电子密度泛函来量化化学概念的努力应该为我们最终开发使用DFT语言的化学反应性理论打开一扇新门。我们将首先概述对其理论框架的当前理解,然后重点介绍化学问题的最新应用,包括异构体和构象稳定性,亲电性和亲核性,强共价和弱非共价相互作用,酸度和碱性,芳香性和抗芳香性以及许多其他特性。利用电子密度泛函来量化化学概念的努力应该为我们最终开发使用DFT语言的化学反应性理论打开一扇新门。我们将首先概述对其理论框架的当前理解,然后重点介绍化学问题的最新应用,包括异构体和构象稳定性,亲电性和亲核性,强共价和弱非共价相互作用,酸度和碱性,芳香性和抗芳香性以及许多其他特性。利用电子密度泛函来量化化学概念的努力应该为我们最终开发使用DFT语言的化学反应性理论打开一扇新门。芳香性和抗芳香性,以及许多其他特性。利用电子密度泛函来量化化学概念的努力应该为我们最终使用DFT语言发展化学反应性理论打开一扇新门。芳香性和抗芳香性,以及许多其他特性。利用电子密度泛函来量化化学概念的努力应该为我们最终开发使用DFT语言的化学反应性理论打开一扇新门。






























 京公网安备 11010802027423号
京公网安备 11010802027423号