Chemical Engineering Journal ( IF 13.3 ) Pub Date : 2019-12-24 , DOI: 10.1016/j.cej.2019.123923
Hanrui Su , Yan Wei , Xiaolei Qu , Chunyang Yu , Qilin Li , Pedro J.J. Alvarez , Mingce Long
|
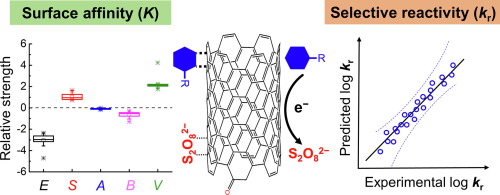
There is increasing interest in persulfate (peroxydisulfate (PDS) and peroxymonosulfate (PMS)) activation by (non-toxic) metal-free catalysts for advanced oxidation of organic contaminants. Nevertheless, the non-radical reaction mechanisms of these catalytic reactions are not well understood. Herein, we provide mechanistic inference on the non-radical reaction pathway of the carbon nanotubes (CNTs)/persulfate system and the associated kinetics. We determined the initial degradation rates of 18 phenolic and 4 aniline compounds at various initial concentrations by commercial multiwalled carbon nanotubes (MWCNTs) and peroxydisulfate (PDS), which followed binary Langmuir−Hinshelwood kinetics. Polyparameter linear free energy relationships (pp-LFERs) and quantitative structure-activity relationships (QSARs) were developed for the equilibrium adsorption constant (K) and the surface reaction rate constant (kr). QSAR analyses for logK suggested that affinity interactions between organics and MWCNTs are mainly related to π-π interactions. The molecular volume term (V) in the pp-LEFRs contributes the most to the interaction, indicating hydrophobic interactions are also significant. QSAR analyses for log kr indicated one-electron oxidation potential (Eox, representing single-electron transfer ability) and polarizability (α) are predominant factors affecting the reactivity of aromatic organics towards MWCNTs/PDS. Additionally, non-radical reactions occur between adsorbed aromatic compounds and adsorbed PDS, indicating that surface interactions and reactions with MWCNTs dominate the overall reaction rates.
中文翻译:
碳纳米管活化的过二硫酸盐体系中苯酚和苯胺反应动力学的机理推断:pp-LFERs和QSAR分析
人们对通过(无毒)无金属催化剂活化过硫酸盐(过氧二硫酸盐(PDS)和过氧单硫酸盐(PMS))进行高级氧化的兴趣日益浓厚。然而,这些催化反应的非自由基反应机理还没有被很好地理解。在本文中,我们提供了对碳纳米管/过硫酸盐体系的非自由基反应途径及其相关动力学的机理推断。我们通过商业化的多壁碳纳米管(MWCNT)和过氧二硫酸盐(PDS)确定了在各种初始浓度下18种酚和4种苯胺化合物的初始降解速率,并遵循了二元Langmuir-Hinshelwood动力学。K)和表面反应速率常数(k r)。对log K的QSAR分析表明,有机物与MWCNT之间的亲和力相互作用主要与π-π相互作用有关。pp-LEFRs中的分子体积项(V)对相互作用的影响最大,表明疏水性相互作用也很重要。QSAR对log k r的分析表明单电子氧化电位(E ox,表示单电子转移能力)和极化率(α)是影响芳族有机物对MWCNTs / PDS的反应性的主要因素。另外,吸附的芳族化合物和吸附的PDS之间会发生非自由基反应,这表明表面反应和与MWCNT的反应主导了整体反应速率。






























 京公网安备 11010802027423号
京公网安备 11010802027423号