当前位置:
X-MOL 学术
›
J. Chem. Eng. Data
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Prediction of Alkanolamine pKa Values by Combined Molecular Dynamics Free Energy Simulations and ab Initio Calculations
Journal of Chemical & Engineering Data ( IF 2.0 ) Pub Date : 2019-12-18 , DOI: 10.1021/acs.jced.9b00927 Javad Noroozi 1 , William R. Smith 2, 3
Journal of Chemical & Engineering Data ( IF 2.0 ) Pub Date : 2019-12-18 , DOI: 10.1021/acs.jced.9b00927 Javad Noroozi 1 , William R. Smith 2, 3
Affiliation
|
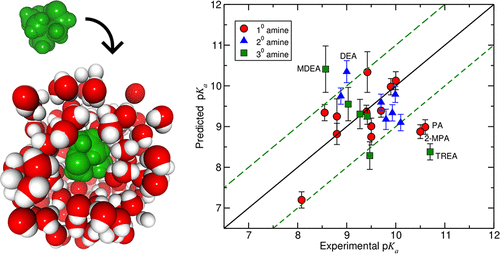
Knowledge of aqueous protonation constants (pKa) of chemical species is of significant importance in CO2 reactive absorption system design. Their theoretical prediction has mainly relied on implicit solvent models, and the performance of explicit solvent simulations based on classical force fields have rarely been studied. In this paper, we report the results of simulations in explicit TIP3P water with the General Amber Force Field (GAFF) and with the SMD continuum solvent method for the deprotonation pKa values of 29 conformationally diverse alkanolamine species commonly used in CO2 capture. In both cases, we employ the Tissandier value for the hydration free energy of the proton (“The proton’s absolute aqueous enthalpy and Gibbs free energy of solvation from cluster–ion solvation data”, Tissandier, M.D. et al., J. Phys. Chem. A, 1998, 102, 7787–7794). The ideal–gas reaction free energies and their uncertainties were obtained from electronic structure calculations using five different compound methods (CBS-QB3, CBS-APNO, G3, G3B3, G4). The hydration free energies of the neutral and protonated forms of the alkanolamines were calculated using the semiempirical AM1-BCC charge method, in addition to several partial atomic charge sets based on the RESP fitting method using electrostatic potentials computed at different ab initio theory/levels in the gas phase as well as in the presence of the solvent reaction field. We incorporated the Galvani surface potential of the ions in the (pKa) calculations. Although the individual species hydration free energies show significant sensitivity to the charge model, the resulting pKa values from different charge models are quite similar. Moreover, we found that the protonated amine hydration free energies show slightly less sensitivity to the partial charge method than in the case of the neutral amine. While the predicted pKa values based on the RESP charges yield reasonable agreement with the experimental data, they are prone to occasional disagreement for molecules of complex geometry. The best performance was achieved using the semiempirical AM1-BCC charges, which showed a mean absolute error of less than 0.73 pKa units in comparison with experimental data. Our results suggest that the AM1-BCC charge method may be used to model electrolyte solutions encountered in the CO2 reactive absorption process.
中文翻译:

结合分子动力学自由能模拟和从头算的预测烷醇胺p K a值
了解化学物种的水质子化常数(p K a)在CO 2反应吸收系统设计中具有重要意义。他们的理论预测主要依靠隐式溶剂模型,并且很少研究基于经典力场的显式溶剂模拟的性能。在本文中,我们报告了使用普通琥珀色力场(GAFF)和SMD连续介质溶剂法在显性TIP3P水中进行去质子化p K a值的模拟结果,该值通常用于CO 2中的29种构象多样的烷醇胺种类捕获。在这两种情况下,我们都使用质子的水合自由能的Tissandier值(“簇离子离子化数据中质子的绝对水焓和Gibbs的溶剂化自由能”,Tissandier,MD等,J。Phys。Chem。一种,1998年,102,7787–7794)。理想气体反应自由能及其不确定性是通过使用五种不同的复合方法(CBS-QB3,CBS-APNO,G3,G3B3,G4)通过电子结构计算获得的。使用半经验AM1-BCC电荷方法计算链烷醇胺的中性和质子化形式的水合自由能,此外还使用基于RESP拟合方法的几个部分原子电荷集,使用在不同的从头算理论/水平计算的静电势。气相以及在存在溶剂反应场的情况下。我们在(p K a)计算中并入了离子的Galvani表面电势。尽管单个物质的水合自由能对电荷模型显示出显着的敏感性,但最终的p K来自不同收费模型的值非常相似。此外,我们发现质子化胺水合自由能显示出比中性胺对部分电荷法更不敏感的灵敏度。尽管基于RESP电荷的预测p K a值与实验数据可以合理地吻合,但对于复杂几何形状的分子而言,它们有时会出现分歧。使用半经验AM1-BCC电荷可达到最佳性能,与实验数据相比,平均电荷误差小于0.73 p K a单位。我们的结果表明,AM1-BCC充电方法可用于模拟CO 2中遇到的电解质溶液 反应吸收过程。
更新日期:2019-12-19
中文翻译:
结合分子动力学自由能模拟和从头算的预测烷醇胺p K a值
了解化学物种的水质子化常数(p K a)在CO 2反应吸收系统设计中具有重要意义。他们的理论预测主要依靠隐式溶剂模型,并且很少研究基于经典力场的显式溶剂模拟的性能。在本文中,我们报告了使用普通琥珀色力场(GAFF)和SMD连续介质溶剂法在显性TIP3P水中进行去质子化p K a值的模拟结果,该值通常用于CO 2中的29种构象多样的烷醇胺种类捕获。在这两种情况下,我们都使用质子的水合自由能的Tissandier值(“簇离子离子化数据中质子的绝对水焓和Gibbs的溶剂化自由能”,Tissandier,MD等,J。Phys。Chem。一种,1998年,102,7787–7794)。理想气体反应自由能及其不确定性是通过使用五种不同的复合方法(CBS-QB3,CBS-APNO,G3,G3B3,G4)通过电子结构计算获得的。使用半经验AM1-BCC电荷方法计算链烷醇胺的中性和质子化形式的水合自由能,此外还使用基于RESP拟合方法的几个部分原子电荷集,使用在不同的从头算理论/水平计算的静电势。气相以及在存在溶剂反应场的情况下。我们在(p K a)计算中并入了离子的Galvani表面电势。尽管单个物质的水合自由能对电荷模型显示出显着的敏感性,但最终的p K来自不同收费模型的值非常相似。此外,我们发现质子化胺水合自由能显示出比中性胺对部分电荷法更不敏感的灵敏度。尽管基于RESP电荷的预测p K a值与实验数据可以合理地吻合,但对于复杂几何形状的分子而言,它们有时会出现分歧。使用半经验AM1-BCC电荷可达到最佳性能,与实验数据相比,平均电荷误差小于0.73 p K a单位。我们的结果表明,AM1-BCC充电方法可用于模拟CO 2中遇到的电解质溶液 反应吸收过程。




















































 京公网安备 11010802027423号
京公网安备 11010802027423号