当前位置:
X-MOL 学术
›
J. Mol. Spectrosc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
The Interplay of VSCF/VCI calculations and Matrix-Isolation IR Spectroscopy - Mid Infrared Spectrum of CH3CH2F and CD3CD2F
Journal of Molecular Spectroscopy ( IF 1.4 ) Pub Date : 2020-01-01 , DOI: 10.1016/j.jms.2019.111224 Dennis F. Dinu , Benjamin Ziegler , Maren Podewitz , Klaus R. Liedl , Thomas Loerting , Hinrich Grothe , Guntram Rauhut
Journal of Molecular Spectroscopy ( IF 1.4 ) Pub Date : 2020-01-01 , DOI: 10.1016/j.jms.2019.111224 Dennis F. Dinu , Benjamin Ziegler , Maren Podewitz , Klaus R. Liedl , Thomas Loerting , Hinrich Grothe , Guntram Rauhut
|
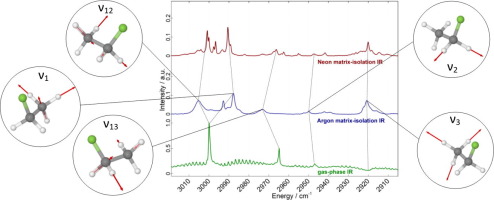
Abstract We present the first matrix-isolation infrared (MI-IR) spectra of CH3CH2F and its isotopologue CD3CD2F in Neon and Argon matrix, together with new gas-phase IR spectra. Extensive vibrational self-consistent field and configuration interaction (VSCF/VCI) calculations are performed, based on an ab initio potential energy surface at ae-CCSD(T)-F12a/cc-pCVTZ-F12 level of electronic structure theory. We encounter an excellent agreement between VCI calculated transitions and the experimental MI-IR and gas-phase IR spectra. Mean absolute deviations are scattering between 1 and 4 cm−1. The interplay of accurate vibrational structure calculations and high-resolution infrared experiments enables unprecedented insights in the CH respectively CD stretch region, providing the first rigorous assignment of the energetically very close ν 1 , ν 12 and ν 13 fundamental transitions.
中文翻译:

VSCF/VCI 计算与基质隔离红外光谱的相互作用 - CH3CH2F 和 CD3CD2F 的中红外光谱
摘要 我们首次展示了氖气和氩气基质中 CH3CH2F 及其同位素体 CD3CD2F 的基质隔离红外 (MI-IR) 光谱,以及新的气相红外光谱。基于 ae-CCSD(T)-F12a/cc-pCVTZ-F12 电子结构理论水平的 ab initio 势能面,进行了广泛的振动自洽场和构型相互作用 (VSCF/VCI) 计算。我们发现 VCI 计算的跃迁与实验 MI-IR 和气相 IR 光谱之间存在极好的一致性。平均绝对偏差在 1 到 4 cm-1 之间发生散射。精确的振动结构计算和高分辨率红外实验的相互作用使得对 CH 和 CD 拉伸区域前所未有的洞察力,提供了能量上非常接近的 ν 1 的第一个严格分配,
更新日期:2020-01-01
中文翻译:
VSCF/VCI 计算与基质隔离红外光谱的相互作用 - CH3CH2F 和 CD3CD2F 的中红外光谱
摘要 我们首次展示了氖气和氩气基质中 CH3CH2F 及其同位素体 CD3CD2F 的基质隔离红外 (MI-IR) 光谱,以及新的气相红外光谱。基于 ae-CCSD(T)-F12a/cc-pCVTZ-F12 电子结构理论水平的 ab initio 势能面,进行了广泛的振动自洽场和构型相互作用 (VSCF/VCI) 计算。我们发现 VCI 计算的跃迁与实验 MI-IR 和气相 IR 光谱之间存在极好的一致性。平均绝对偏差在 1 到 4 cm-1 之间发生散射。精确的振动结构计算和高分辨率红外实验的相互作用使得对 CH 和 CD 拉伸区域前所未有的洞察力,提供了能量上非常接近的 ν 1 的第一个严格分配,




















































 京公网安备 11010802027423号
京公网安备 11010802027423号