当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Molecular Dynamic Simulations of Clathrate Hydrate Anomalous Preservation: The Effect of Coating Clathrate Hydrate Phases
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2019-11-15 , DOI: 10.1021/acs.jpcc.9b07769 Parisa Naeiji 1 , Tom K. Woo 1 , Saman Alavi 1, 2 , John A. Ripmeester 2
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2019-11-15 , DOI: 10.1021/acs.jpcc.9b07769 Parisa Naeiji 1 , Tom K. Woo 1 , Saman Alavi 1, 2 , John A. Ripmeester 2
Affiliation
|
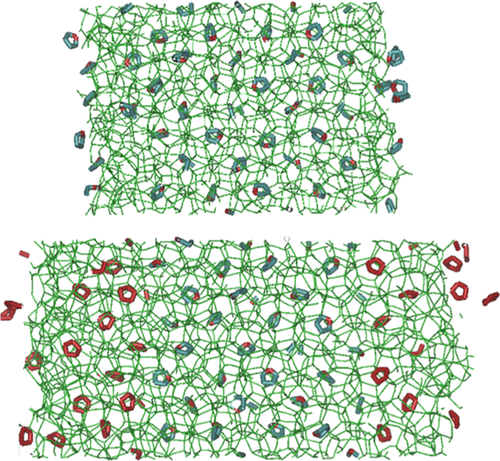
In this work, the effect of cyclopentane (CP) clathrate hydrate on the anomalous preservation of tetrahydrofuran (THF) hydrate under conditions outside its stability region is studied by using molecular dynamics simulations. The decompositions of pure structure II THF and CP clathrate hydrate, and also THF hydrate coated by CP hydrate, all with outer (001) surfaces exposed to vacuum, were simulated at different temperatures and characterized by the potential energy, the F3 order parameter, and visual inspection of snapshots of the hydrate system at different times. The upper bounding melting points of THF and the CP hydrate with the employed force fields were predicted to be 270 and 290 K, respectively, which were close to the experimental values of 277.5 and 281 K. To study the origins of anomalous preservation and superheating effects in hydrates, we placed layers of the higher-decomposition point CP hydrate as a coating on bulk THF hydrate to study the possible superheating of the THF hydrate. Whereas the pure THF hydrate melted at 270 K after a simulation time of about 50 ns, with the CP hydrate layer coating, the THF hydrate in the simulations did not dissociate at 290 K, corresponding to a superheating temperature of 20 K, up to a simulation time of 120 ns. Upon coating with the CP hydrate, the decomposition of the THF hydrate is transformed from a heterogeneous mechanism at the hydrate–vacuum or hydrate–water interface to a homogeneous mechanism which leads to superheating of the THF hydrate phase. The dissociation of the THF and the CP hydrate layers occurs in a stepwise fashion perpendicular to the hydrate interface from the outer to inner layers, similar to the dissociation of the structure I methane hydrate previously studied.
中文翻译:

包合物水合物异常保存的分子动力学模拟:包合物包合物水合物相的影响
在这项工作中,通过分子动力学模拟研究了环戊烷(CP)笼形水合物对四氢呋喃(THF)水合物在其稳定区域外的异常保存的影响。在不同温度下模拟纯结构II THF和CP笼形水合物的分解,以及CP水合物包被的THF水合物的分解,所有外表面(001)都暴露于真空中,并以势能F 3为特征。顺序参数,以及在不同时间目视检查水合物系统的快照。预测了使用力场时THF和CP水合物的上限熔点分别为270 K和290 K,接近实验值277.5和281K。研究异常保存和过热效应的起源在水合物中,我们将较高分解点CP水合物的层作为主体THF水合物的涂层,以研究THF水合物的可能过热。在约50 ns的模拟时间后,纯THF水合物在270 K时熔化,并带有CP水合物层涂层,而在模拟条件下,THF水合物在290 K时未解离,这对应于20 K的过热温度,直至仿真时间为120 ns。用CP水合物包衣后,THF水合物的分解从水合物-真空或水合物-水界面的非均质机理转变为均质机理,导致THF水合物相过热。THF和CP水合物层的离解垂直于水合物界面从外层到内层以逐步方式发生,类似于先前研究的结构I甲烷水合物的离解。
更新日期:2019-11-17
中文翻译:
包合物水合物异常保存的分子动力学模拟:包合物包合物水合物相的影响
在这项工作中,通过分子动力学模拟研究了环戊烷(CP)笼形水合物对四氢呋喃(THF)水合物在其稳定区域外的异常保存的影响。在不同温度下模拟纯结构II THF和CP笼形水合物的分解,以及CP水合物包被的THF水合物的分解,所有外表面(001)都暴露于真空中,并以势能F 3为特征。顺序参数,以及在不同时间目视检查水合物系统的快照。预测了使用力场时THF和CP水合物的上限熔点分别为270 K和290 K,接近实验值277.5和281K。研究异常保存和过热效应的起源在水合物中,我们将较高分解点CP水合物的层作为主体THF水合物的涂层,以研究THF水合物的可能过热。在约50 ns的模拟时间后,纯THF水合物在270 K时熔化,并带有CP水合物层涂层,而在模拟条件下,THF水合物在290 K时未解离,这对应于20 K的过热温度,直至仿真时间为120 ns。用CP水合物包衣后,THF水合物的分解从水合物-真空或水合物-水界面的非均质机理转变为均质机理,导致THF水合物相过热。THF和CP水合物层的离解垂直于水合物界面从外层到内层以逐步方式发生,类似于先前研究的结构I甲烷水合物的离解。






























 京公网安备 11010802027423号
京公网安备 11010802027423号