Bone Research ( IF 14.3 ) Pub Date : 2019-11-05 , DOI: 10.1038/s41413-019-0070-y
Courtney M Mazur 1, 2 , Jonathon J Woo 1 , Cristal S Yee 1 , Aaron J Fields 1 , Claire Acevedo 1, 3 , Karsyn N Bailey 1, 2 , Serra Kaya 1 , Tristan W Fowler 1 , Jeffrey C Lotz 1, 2 , Alexis Dang 1, 4 , Alfred C Kuo 1, 4 , Thomas P Vail 1 , Tamara Alliston 1, 2
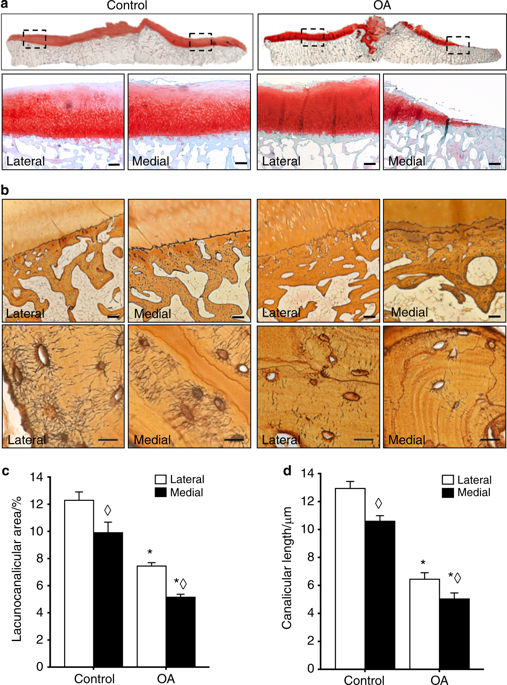
|
Osteoarthritis (OA), long considered a primary disorder of articular cartilage, is commonly associated with subchondral bone sclerosis. However, the cellular mechanisms responsible for changes to subchondral bone in OA, and the extent to which these changes are drivers of or a secondary reaction to cartilage degeneration, remain unclear. In knee joints from human patients with end-stage OA, we found evidence of profound defects in osteocyte function. Suppression of osteocyte perilacunar/canalicular remodeling (PLR) was most severe in the medial compartment of OA subchondral bone, with lower protease expression, diminished canalicular networks, and disorganized and hypermineralized extracellular matrix. As a step toward evaluating the causality of PLR suppression in OA, we ablated the PLR enzyme MMP13 in osteocytes while leaving chondrocytic MMP13 intact, using Cre recombinase driven by the 9.6-kb DMP1 promoter. Not only did osteocytic MMP13 deficiency suppress PLR in cortical and subchondral bone, but it also compromised cartilage. Even in the absence of injury, osteocytic MMP13 deficiency was sufficient to reduce cartilage proteoglycan content, change chondrocyte production of collagen II, aggrecan, and MMP13, and increase the incidence of cartilage lesions, consistent with early OA. Thus, in humans and mice, defects in PLR coincide with cartilage defects. Osteocyte-derived MMP13 emerges as a critical regulator of cartilage homeostasis, likely via its effects on PLR. Together, these findings implicate osteocytes in bone-cartilage crosstalk in the joint and suggest a causal role for suppressed perilacunar/canalicular remodeling in osteoarthritis.
中文翻译:
骨细胞功能障碍通过 MMP13 依赖性抑制软骨下骨稳态促进骨关节炎
骨关节炎(OA)长期以来被认为是关节软骨的原发性疾病,通常与软骨下骨硬化有关。然而,导致 OA 软骨下骨变化的细胞机制,以及这些变化在多大程度上是软骨退变的驱动因素或继发反应,仍不清楚。在患有终末期骨关节炎的人类患者的膝关节中,我们发现了骨细胞功能严重缺陷的证据。骨细胞周围/小管重塑(PLR)的抑制在OA软骨下骨的内侧隔室中最为严重,蛋白酶表达较低,小管网络减少,细胞外基质紊乱和矿化过度。作为评估 OA 中 PLR 抑制因果关系的一步,我们使用由 9.6-kb DMP1 启动子驱动的 Cre 重组酶消除了骨细胞中的 PLR 酶 MMP13,同时保持软骨细胞 MMP13 完整。骨细胞 MMP13 缺乏不仅会抑制皮质骨和软骨下骨中的 PLR,而且还会损害软骨。即使没有损伤,骨细胞 MMP13 缺乏也足以减少软骨蛋白聚糖含量,改变软骨细胞生成 II 型胶原蛋白、聚集蛋白聚糖和 MMP13,并增加软骨损伤的发生率,这与早期 OA 一致。因此,在人类和小鼠中,PLR 缺陷与软骨缺陷一致。骨细胞来源的 MMP13 可能通过其对 PLR 的影响而成为软骨稳态的关键调节因子。总之,这些发现表明骨细胞参与关节中的骨-软骨串扰,并表明骨关节炎中腔周/小管重塑受到抑制的因果作用。






























 京公网安备 11010802027423号
京公网安备 11010802027423号