当前位置:
X-MOL 学术
›
J. Chin. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
A profound density functional theory study to unravel the spectroscopic and molecular properties of two Flavanols differing in α‐pyrone ring position
Journal of the Chinese Chemical Society ( IF 1.6 ) Pub Date : 2019-11-04 , DOI: 10.1002/jccs.201900334 Muhammad Ali Hashmi 1 , Umar Farooq 2, 3 , Syeda Sidra Bibi 2 , Sadia Naz 2, 4 , Hong‐Guang Xu 3 , Basim H. Asghar 5 , Yahia Nasser Mabkhot 6 , Abdulrahman Alsayari 7 , Abdullatif Bin Muhsinah 7 , Ayesha Khan 8
Journal of the Chinese Chemical Society ( IF 1.6 ) Pub Date : 2019-11-04 , DOI: 10.1002/jccs.201900334 Muhammad Ali Hashmi 1 , Umar Farooq 2, 3 , Syeda Sidra Bibi 2 , Sadia Naz 2, 4 , Hong‐Guang Xu 3 , Basim H. Asghar 5 , Yahia Nasser Mabkhot 6 , Abdulrahman Alsayari 7 , Abdullatif Bin Muhsinah 7 , Ayesha Khan 8
Affiliation
|
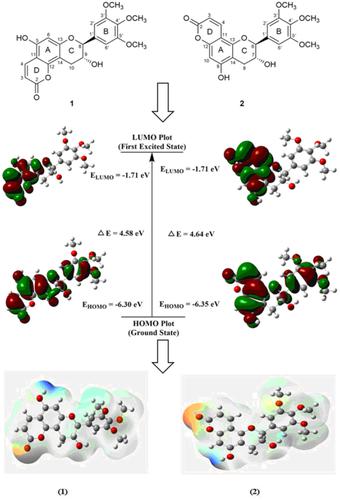
A comprehensive theoretical model was designed for two new flavanols that have been reported from Glycosmis pentaphylla, differing in the placement of α‐pyrone ring. The density functional theory (DFT) approach was utilized for computing different properties of these compounds to validate the experimental findings and stereochemical assignments. Electronic properties, geometric parameters, frontier molecular orbitals (FMOs), molecular electrostatic potential (MESP), and natural bond orbital analysis were performed for the first time at the PBE0‐D3BJ/def2‐TZVP level of theory for the compounds under study. The simulated vibrational frequencies for compounds 1 and 2 were computed and compared with the experimental results. nuclear magnetic resonance (NMR) (1H and 13C) chemical shift values were computed at the PBE0‐D3BJ/def2‐TZVP/SMDDMSO level of theory and showed a very good agreement with the experimental results for both the compounds. The electronic circular dichroism (ECD) and ultraviolet–visible (UV) spectra for both the compounds were obtained using time‐dependent DFT in methanol, whose results exhibited excellent correlation with experimental data. The intermolecular interaction effect on geometric parameters, vibrational frequencies, and electronic properties were studied for the first time.
中文翻译:

深入的密度泛函理论研究,揭示了两种不同α-吡喃酮环位置的黄烷醇的光谱和分子性质
全面的理论模型是专为已经从报道的两个新的黄烷醇山小橘,在安置不同的α吡喃酮环。密度泛函理论(DFT)方法用于计算这些化合物的不同性质,以验证实验结果和立体化学分配。对于所研究化合物,首次以PBE0-D3BJ / def2-TZVP的理论水平进行了电子性质,几何参数,前沿分子轨道(FMO),分子静电势(MESP)和自然键轨道分析。化合物1和2的模拟振动频率计算并与实验结果进行比较。核磁共振(NMR)(1 H和13 C)的化学位移值是在PBE0-D3BJ / def2-TZVP / SMD DMSO的理论水平上计算得出的,与两种化合物的实验结果都非常吻合。两种化合物的电子圆二色性(ECD)和紫外-可见(UV)光谱是在甲醇中使用时间依赖性DFT获得的,其结果与实验数据具有极好的相关性。首次研究了分子间相互作用对几何参数,振动频率和电子性质的影响。
更新日期:2019-11-04
中文翻译:
深入的密度泛函理论研究,揭示了两种不同α-吡喃酮环位置的黄烷醇的光谱和分子性质
全面的理论模型是专为已经从报道的两个新的黄烷醇山小橘,在安置不同的α吡喃酮环。密度泛函理论(DFT)方法用于计算这些化合物的不同性质,以验证实验结果和立体化学分配。对于所研究化合物,首次以PBE0-D3BJ / def2-TZVP的理论水平进行了电子性质,几何参数,前沿分子轨道(FMO),分子静电势(MESP)和自然键轨道分析。化合物1和2的模拟振动频率计算并与实验结果进行比较。核磁共振(NMR)(1 H和13 C)的化学位移值是在PBE0-D3BJ / def2-TZVP / SMD DMSO的理论水平上计算得出的,与两种化合物的实验结果都非常吻合。两种化合物的电子圆二色性(ECD)和紫外-可见(UV)光谱是在甲醇中使用时间依赖性DFT获得的,其结果与实验数据具有极好的相关性。首次研究了分子间相互作用对几何参数,振动频率和电子性质的影响。
































 京公网安备 11010802027423号
京公网安备 11010802027423号