当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Molecular Dynamics Simulations of Crystal Nucleation from Solution at Constant Chemical Potential.
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2019-11-12 , DOI: 10.1021/acs.jctc.9b00795 Tarak Karmakar 1, 2 , Pablo M Piaggi 1, 2 , Michele Parrinello 1, 2
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2019-11-12 , DOI: 10.1021/acs.jctc.9b00795 Tarak Karmakar 1, 2 , Pablo M Piaggi 1, 2 , Michele Parrinello 1, 2
Affiliation
|
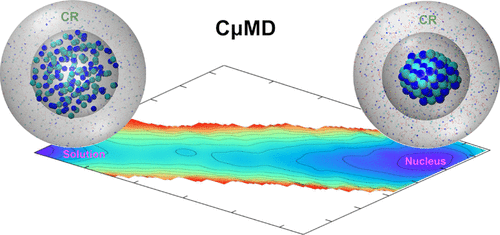
A widespread method of crystal preparation is to precipitate it from a supersaturated solution. In such a process, control of solution concentration is of paramount importance. The nucleation process, polymorph selection, and crystal habits depend crucially on this thermodynamic parameter. When performing molecular dynamics simulations with a fixed number of molecules in the canonical ensemble, crystal growth is accompanied by a decrease in the solution concentration. This modification of the thermodynamic condition leads to significant artifacts. Inspired by the recent development of the constant chemical potential molecular dynamics simulation method by Perego et al. [ J. Chem. Phys. 2015 , 142 , 144113 ] , we develop a spherical variant of it to study nucleation from solution. Our method allows determining the crystal nucleus size and nucleation rates at constant supersaturation. As an example, we study the homogeneous nucleation of sodium chloride from its supersaturated aqueous solution.
中文翻译:

恒定化学势下溶液中晶体成核的分子动力学模拟。
广泛的晶体制备方法是从过饱和溶液中沉淀出来。在这样的过程中,溶液浓度的控制至关重要。成核过程,多晶型物的选择和晶体习性主要取决于此热力学参数。在规范集合中使用固定数量的分子执行分子动力学模拟时,晶体生长伴随溶液浓度的降低。热力学条件的这种改变导致明显的伪像。受到Perego等人最近开发的恒定化学势分子动力学模拟方法的启发。[J. Chem。物理 2015,142,144113],我们开发了它的球形变体,以研究溶液中的形核。我们的方法允许在恒定的过饱和状态下确定晶核的大小和成核速率。例如,我们研究了氯化钠从其过饱和水溶液中的均匀成核。
更新日期:2019-11-13
中文翻译:
恒定化学势下溶液中晶体成核的分子动力学模拟。
广泛的晶体制备方法是从过饱和溶液中沉淀出来。在这样的过程中,溶液浓度的控制至关重要。成核过程,多晶型物的选择和晶体习性主要取决于此热力学参数。在规范集合中使用固定数量的分子执行分子动力学模拟时,晶体生长伴随溶液浓度的降低。热力学条件的这种改变导致明显的伪像。受到Perego等人最近开发的恒定化学势分子动力学模拟方法的启发。[J. Chem。物理 2015,142,144113],我们开发了它的球形变体,以研究溶液中的形核。我们的方法允许在恒定的过饱和状态下确定晶核的大小和成核速率。例如,我们研究了氯化钠从其过饱和水溶液中的均匀成核。





























 京公网安备 11010802027423号
京公网安备 11010802027423号