当前位置:
X-MOL 学术
›
J. Phys. Chem. B
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Singlet Energy Transfer in Anthracene-Porphyrin Complexes: Mechanism, Geometry, and Implications for Intramolecular Photon Upconversion.
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2019-11-07 , DOI: 10.1021/acs.jpcb.9b07991
Fredrik Edhborg 1 , Betül Küçüköz 1 , Victor Gray 1 , Bo Albinsson 1
The Journal of Physical Chemistry B ( IF 2.8 ) Pub Date : 2019-11-07 , DOI: 10.1021/acs.jpcb.9b07991
Fredrik Edhborg 1 , Betül Küçüköz 1 , Victor Gray 1 , Bo Albinsson 1
Affiliation
|
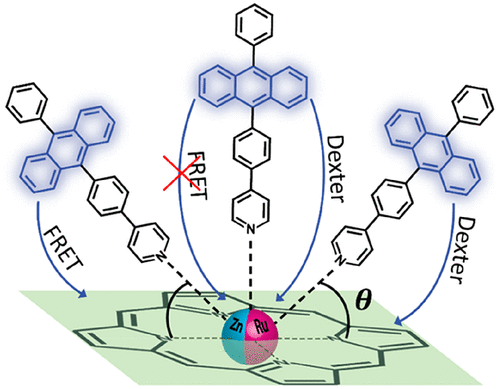
In this work we show that the mechanism for singlet excitation energy transfer (SET) in coordination complexes changes upon changing a single atom. SET is governed by two different mechanisms; Förster resonance energy transfer (FRET) based on Coulombic, through-space interactions, or Dexter energy transfer relying on exchange, through-bond interactions. On the basis of time-resolved fluorescence and transient absorption measurements, we conduct a mechanistic study of SET from a set of photoexcited anthracene donors to axially coordinated porphyrin acceptors, revealing the effect of coordination geometry and a very profound effect of the porphyrin central metal atom. We found that FRET is the dominating mechanism of SET for complexes with zinc-octaethylporphyrin (ZnOEP) as the acceptor, while Dexter energy transfer is the dominating mechanism of SET in a corresponding ruthenium complex (RuOEP). In addition, by analyzing the coordination geometry of the complexes and its temperature dependence, the binding angle potential energy of axially coordinated porphyrin complexes could be estimated. The results of this study are of fundamental importance and are discussed with respect to the consequences for developing intramolecular triplet-triplet annihilation photon upconversion in coordination complexes.
中文翻译:

蒽-卟啉配合物中的单线态能量转移:分子内光子上转换的机理,几何学和意义。
在这项工作中,我们表明配位络合物中单重态激发能转移(SET)的机制在改变单个原子时发生变化。SET由两种不同的机制控制;基于库仑空间相互作用的Förster共振能量转移(FRET),或依赖交换,贯穿键相互作用的Dexter能量转移。基于时间分辨的荧光和瞬态吸收测量,我们从一组光激发的蒽供体到轴向配位的卟啉受体进行了SET的机理研究,揭示了配位几何学的影响以及卟啉中心金属原子的非常深刻的影响。我们发现,对于以八乙基卟啉锌(ZnOEP)为受体的配合物,FRET是SET的主要机制,而Dexter的能量转移是相应钌络合物(RuOEP)中SET的主要机制。此外,通过分析配合物的配位几何形状及其温度依赖性,可以估算轴向配位的卟啉配合物的结合角势能。这项研究的结果具有根本的重要性,并讨论了在配位配合物中发展分子内三重态-三重态an灭光子上转换的后果。
更新日期:2019-11-08
中文翻译:
蒽-卟啉配合物中的单线态能量转移:分子内光子上转换的机理,几何学和意义。
在这项工作中,我们表明配位络合物中单重态激发能转移(SET)的机制在改变单个原子时发生变化。SET由两种不同的机制控制;基于库仑空间相互作用的Förster共振能量转移(FRET),或依赖交换,贯穿键相互作用的Dexter能量转移。基于时间分辨的荧光和瞬态吸收测量,我们从一组光激发的蒽供体到轴向配位的卟啉受体进行了SET的机理研究,揭示了配位几何学的影响以及卟啉中心金属原子的非常深刻的影响。我们发现,对于以八乙基卟啉锌(ZnOEP)为受体的配合物,FRET是SET的主要机制,而Dexter的能量转移是相应钌络合物(RuOEP)中SET的主要机制。此外,通过分析配合物的配位几何形状及其温度依赖性,可以估算轴向配位的卟啉配合物的结合角势能。这项研究的结果具有根本的重要性,并讨论了在配位配合物中发展分子内三重态-三重态an灭光子上转换的后果。
































 京公网安备 11010802027423号
京公网安备 11010802027423号