当前位置:
X-MOL 学术
›
Appl. Surf. Sci.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
不同PAM结构单元在高岭石(001)表面的吸附:密度泛函理论研究
Applied Surface Science ( IF 6.3 ) Pub Date : 2020-02-01 , DOI: 10.1016/j.apsusc.2019.144324 Bao Ren , Fanfei Min , Lingyun Liu , Jun Chen , Chunfu Liu , Kai Lv
Applied Surface Science ( IF 6.3 ) Pub Date : 2020-02-01 , DOI: 10.1016/j.apsusc.2019.144324 Bao Ren , Fanfei Min , Lingyun Liu , Jun Chen , Chunfu Liu , Kai Lv
|
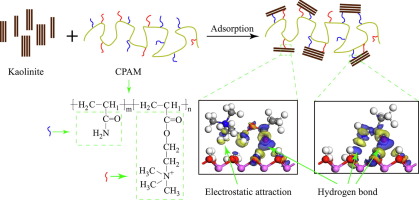
摘要 本研究的目的是探索聚丙烯酰胺 (PAM) 如何与高岭石 (0 0 1) 表面相互作用的吸附机制。从 PAM 的结构发展出三种结构单元模型(P-AM、P-AA 和 P-DAC)。用密度泛函理论(DFT)计算吸附能以确定PAM结构单元在高岭石(0 0 1)表面的最佳吸附体系。从最佳吸附系统的Mulliken布居分析、电子密度差、电子态密度和电子局域函数,推导出各吸附系统的吸附机理并理解它们的差异。结果表明,三个PAM结构单元的这些反应位点是O原子。P-AM和P-AA可以通过形成氢键吸附在高岭石(0 0 1)表面。使用 P-DAC 的高岭石 (0 0 1) 表面的相互作用由氢键和静电引力驱动,同时确定静电引力作为主要作用。吸附在高岭石 (0 0 1) 表面的 P-AM 的氢键强度高于 P-DAC 和 P-AA。然而,吸附在高岭石 (0 0 1) 表面的 PAM 结构单元的吸附相互作用顺序是 P-DAC > P-AM > P-AA。吸附在高岭石 (0 0 1) 表面的 P-AM 的氢键强度高于 P-DAC 和 P-AA。然而,吸附在高岭石 (0 0 1) 表面的 PAM 结构单元的吸附相互作用顺序是 P-DAC > P-AM > P-AA。吸附在高岭石 (0 0 1) 表面的 P-AM 的氢键强度高于 P-DAC 和 P-AA。然而,吸附在高岭石 (0 0 1) 表面的 PAM 结构单元的吸附相互作用顺序是 P-DAC > P-AM > P-AA。

"点击查看英文标题和摘要"
更新日期:2020-02-01
"点击查看英文标题和摘要"
































 京公网安备 11010802027423号
京公网安备 11010802027423号