当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Machine Learning Configuration Interaction for ab Initio Potential Energy Curves
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2019-10-30 , DOI: 10.1021/acs.jctc.9b00828 Jeremy P. Coe 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2019-10-30 , DOI: 10.1021/acs.jctc.9b00828 Jeremy P. Coe 1
Affiliation
|
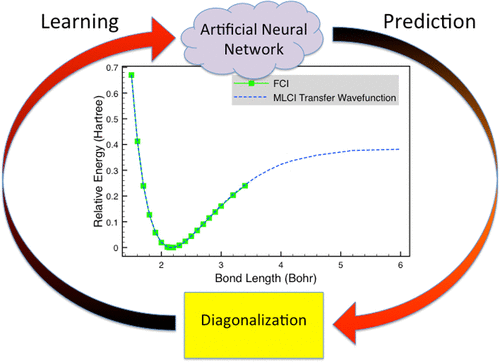
The concept of machine learning configuration interaction (MLCI) (J. Chem. Theory Comput. 2018, 14, 5739), where an artificial neural network (ANN) learns on the fly to select important configurations, is further developed so that accurate ab initio potential energy curves can be efficiently calculated. This development includes employing the artificial neural network also as a hash function for the efficient deletion of duplicates on the fly so that the single and double space does not need to be stored and this barrier to scalability is removed. In addition, configuration state functions are introduced into the approach so that pure spin states are guaranteed, and the transferability of data between geometries is exploited. This improved approach is demonstrated on potential energy curves for the nitrogen molecule, water, and carbon monoxide. The results are compared with full configuration interaction values, when available, and different transfer protocols are investigated. It is shown that, for all of the considered systems, accurate potential energy curves can now be efficiently computed with MLCI. For the potential curves of N2 and CO, MLCI can achieve lower errors than stochastically selecting configurations while also using substantially less processor hours.
中文翻译:

从头算起势能曲线的机器学习配置交互
机器学习配置交互(MLCI)的概念(J.Chem.Theory Comput。2018,14,5739),进一步开发了人工神经网络(ANN)实时学习以选择重要的配置,以便可以有效地计算出准确的从头算势能曲线。此开发包括将人工神经网络也用作哈希函数,以快速有效地删除重复项,从而无需存储单个和双重空间,并且消除了可伸缩性的这一障碍。另外,将配置状态函数引入该方法,从而确保纯自旋状态,并利用几何之间的数据可传递性。在氮分子,水和一氧化碳的势能曲线上证明了这种改进的方法。将结果与完整的配置交互值(如果有)进行比较,并研究了不同的传输协议。结果表明,对于所有考虑的系统,现在都可以使用MLCI有效地计算出准确的势能曲线。对于N的电位曲线如图2和CO所示,与随机选择配置相比,MLCI可以实现更低的错误,同时还可以使用更少的处理器时间。
更新日期:2019-10-30
中文翻译:
从头算起势能曲线的机器学习配置交互
机器学习配置交互(MLCI)的概念(J.Chem.Theory Comput。2018,14,5739),进一步开发了人工神经网络(ANN)实时学习以选择重要的配置,以便可以有效地计算出准确的从头算势能曲线。此开发包括将人工神经网络也用作哈希函数,以快速有效地删除重复项,从而无需存储单个和双重空间,并且消除了可伸缩性的这一障碍。另外,将配置状态函数引入该方法,从而确保纯自旋状态,并利用几何之间的数据可传递性。在氮分子,水和一氧化碳的势能曲线上证明了这种改进的方法。将结果与完整的配置交互值(如果有)进行比较,并研究了不同的传输协议。结果表明,对于所有考虑的系统,现在都可以使用MLCI有效地计算出准确的势能曲线。对于N的电位曲线如图2和CO所示,与随机选择配置相比,MLCI可以实现更低的错误,同时还可以使用更少的处理器时间。
































 京公网安备 11010802027423号
京公网安备 11010802027423号