当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Molecular Dynamics of Graphene–Electrolyte Interface: Interfacial Solution Structure and Molecular Diffusion
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2019-10-17 , DOI: 10.1021/acs.jpcc.9b07487
Jan Dočkal 1 , Filip Moučka 1 , Martin Lísal 1, 2
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2019-10-17 , DOI: 10.1021/acs.jpcc.9b07487
Jan Dočkal 1 , Filip Moučka 1 , Martin Lísal 1, 2
Affiliation
|
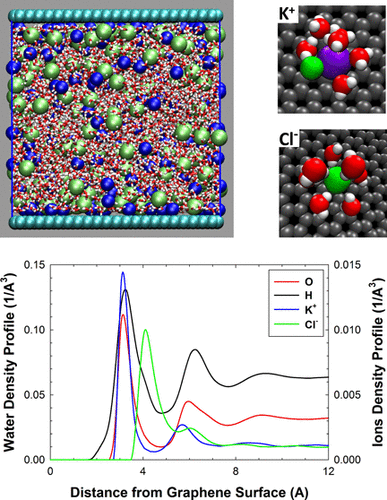
Graphene-based applications often take place in aqueous environments, and they benefit from a molecular-level understanding of aqueous salt solutions in contact with graphene surfaces under different conditions. We study the aqueous solutions of electrolytes (LiCl, NaCl, KCl, MgCl2, and CaCl2) near the interface with a graphene sheet using classical molecular simulations. In order to model the graphene–ion interactions accurately, we use the effective polarizable model of Williams et al. ( J. Phys. Chem. Lett. 2017, 8, 703). In order to thoroughly characterize the solution structure at the graphene surface, in addition to standard structural properties, we employ our novel intermolecular bond definition based on the spatial distribution functions, which provides numbers of water–water and water–ion intermolecular bonds per water molecule and number of water molecules per ion as functions of the distance from the graphene surface in a completely self-consistent manner. This thus allows summations of the bonds and quantitative comparisons of the bonds between different species in the solution. Our analysis shows that the interfacial structure exhibits a competition between strong water structuring, formation of ion dense adsorption layers, and strong hydrogen and ion–water bonds in the solution; what is particularly interesting are the observed charge compensation and the mutual symmetries of intermolecular bonding. Finally, we evaluate the lateral mobility of water and ions separately in the interfacial and bulk regions, showing significant reduction of the dynamics of both the water and the ions in the interfacial region compared to the bulk phase.
中文翻译:

石墨烯-电解质界面的分子动力学:界面溶液结构和分子扩散
基于石墨烯的应用通常发生在水性环境中,并且它们受益于在不同条件下与石墨烯表面接触的盐水溶液的分子水平的理解。我们使用经典的分子模拟研究了与石墨烯片界面附近的电解质水溶液(LiCl,NaCl,KCl,MgCl 2和CaCl 2)。为了准确地建模石墨烯-离子相互作用,我们使用了Williams等人的有效极化模型。(J.物理学。化学快报。 2017,8(703)。为了全面表征石墨烯表面的溶液结构,除了标准的结构特性外,我们还使用了基于空间分布函数的新颖分子间键定义,该定义提供了每个水分子中水-水和水-离子分子间键的数量以及每个离子中水分子的数量以完全自洽的方式表示为与石墨烯表面的距离的函数。因此,这允许溶液中不同种类之间的键求和以及键的定量比较。我们的分析表明,界面结构在强水结构化,离子致密吸附层的形成与溶液中强氢键和离子水键之间存在竞争。特别有趣的是观察到的电荷补偿和分子间键合的相互对称性。最后,我们分别评估了界面区域和主体区域中水和离子的横向迁移率,显示出与主体相相比,界面区域中的水和离子的动力学显着降低。
更新日期:2019-10-17
中文翻译:
石墨烯-电解质界面的分子动力学:界面溶液结构和分子扩散
基于石墨烯的应用通常发生在水性环境中,并且它们受益于在不同条件下与石墨烯表面接触的盐水溶液的分子水平的理解。我们使用经典的分子模拟研究了与石墨烯片界面附近的电解质水溶液(LiCl,NaCl,KCl,MgCl 2和CaCl 2)。为了准确地建模石墨烯-离子相互作用,我们使用了Williams等人的有效极化模型。(J.物理学。化学快报。 2017,8(703)。为了全面表征石墨烯表面的溶液结构,除了标准的结构特性外,我们还使用了基于空间分布函数的新颖分子间键定义,该定义提供了每个水分子中水-水和水-离子分子间键的数量以及每个离子中水分子的数量以完全自洽的方式表示为与石墨烯表面的距离的函数。因此,这允许溶液中不同种类之间的键求和以及键的定量比较。我们的分析表明,界面结构在强水结构化,离子致密吸附层的形成与溶液中强氢键和离子水键之间存在竞争。特别有趣的是观察到的电荷补偿和分子间键合的相互对称性。最后,我们分别评估了界面区域和主体区域中水和离子的横向迁移率,显示出与主体相相比,界面区域中的水和离子的动力学显着降低。






























 京公网安备 11010802027423号
京公网安备 11010802027423号