当前位置:
X-MOL 学术
›
Eur. J. Med. Chem.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Design, synthesis and biological evaluation of N-(3-(1H-tetrazol-1-yl)phenyl)isonicotinamide derivatives as novel xanthine oxidase inhibitors.
European Journal of Medicinal Chemistry ( IF 6.0 ) Pub Date : 2019-09-18 , DOI: 10.1016/j.ejmech.2019.111717 Ting-Jian Zhang 1 , Yi Zhang 1 , Shun Tu 1 , Yu-Hang Wu 1 , Zhen-Hao Zhang 1 , Fan-Hao Meng 1
European Journal of Medicinal Chemistry ( IF 6.0 ) Pub Date : 2019-09-18 , DOI: 10.1016/j.ejmech.2019.111717 Ting-Jian Zhang 1 , Yi Zhang 1 , Shun Tu 1 , Yu-Hang Wu 1 , Zhen-Hao Zhang 1 , Fan-Hao Meng 1
Affiliation
|
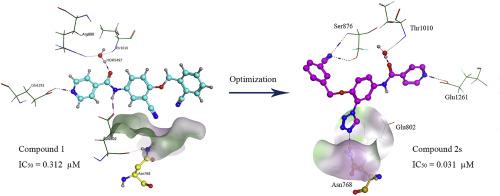
In our previous study, we reported a series of N-phenylisonicotinamide derivatives as novel xanthine oxidase (XO) inhibitors and identified N-(3-cyano-4-((2-cyanobenzyl)oxy)phenyl)isonicotinamide (compound 1) as the most potent one with an IC50 value of 0.312 μM. To further optimize the structure and improve the potency, a structure-based drug design (SBDD) strategy was performed to construct the missing H-bond between the small molecule and the Asn768 residue of XO. We introduced a tetrazole moiety at the 3'-position of the phenyl to serve as an H-bond acceptor and obtained a series of N-(3-(1H-tetrazol-1-yl)phenyl)isonicotinamide derivatives (2a-t and 6-8). Besides, to investigate the influence of the amide-reversal, some N-(pyridin-4-yl)-3-(1H-tetrazol-1-yl)benzamide derivatives (3c, 3e, 3i, 3k and 3u) were also synthesized and evaluated. Biological evaluation and structure-activity relationship analysis demonstrated that the 3'-(1H-tetrazol-1-yl) moiety was an excellent fragment for the N-phenylisonicotinamide scaffold; a substituted benzyloxy, especially, an m-cyanobenzyloxy (e.g., 2s), linking at the 4'-position was welcome for the potency; and the amide-reversal could damage the potency, so maintenance of the N-phenylisonicotinamide scaffold was essential. In summary, starting from compound 1, the SBDD effort successfully identified a promising XO inhibitor 2s (IC50 = 0.031 μM), with a 10-fold gain in potency. Its potency was very close to the positive control topiroxostat (IC50 = 0.021 μM). A Lineweaver-Burk plot indicated that compound 2s acted as a mixed-type XO inhibitor. Molecular docking and molecular dynamics simulations revealed that the tetrazole moiety could occupy the Asn768-sub-pocket with N-4 atom accepting an H-bond from the Asn768 residue, as expected.
中文翻译:

N-(3-(1H-四唑-1-基)苯基)异烟酰胺衍生物作为黄嘌呤氧化酶抑制剂的设计,合成及生物学评价。
在我们先前的研究中,我们报道了一系列N-苯基异烟酰胺衍生物作为新型黄嘌呤氧化酶(XO)抑制剂,并确定了N-(3-氰基-4-((2-氰基苄氧基)氧基)苯基)异烟酰胺(化合物1)是最有效的一种,IC50值为0.312μM。为了进一步优化结构并提高效能,进行了基于结构的药物设计(SBDD)策略,以构建小分子与XO的Asn768残基之间缺失的H键。我们在苯基的3'位引入一个四唑部分作为H键受体,并获得了一系列N-(3-(1H-四唑-1-基)苯基)异烟酰胺衍生物(2a-t和6-8)。此外,为了研究酰胺逆转的影响,还合成了一些N-(吡啶-4-基)-3-(1H-四唑-1-基)苯甲酰胺衍生物(3c,3e,3i,3k和3u)。并进行评估。生物学评估和结构-活性关系分析表明,3'-(1H-四唑-1-基)部分是N-苯基异烟酰胺骨架的极好片段。为了效力,欢迎在4′-位连接的取代的苄氧基,特别是间-氰基苄氧基(例如2s);而且酰胺的逆转可能会破坏药效,因此维持N-苯基异烟酰胺支架是必不可少的。总而言之,从化合物1开始,SBDD的努力成功鉴定出了有希望的XO抑制剂2s(IC50 = 0.031μM),效力提高了10倍。它的效力非常接近于阳性对照托吡司他(IC50 = 0.021μM)。Lineweaver-Burk图表明化合物2s充当了混合型XO抑制剂。
更新日期:2019-09-18
中文翻译:
N-(3-(1H-四唑-1-基)苯基)异烟酰胺衍生物作为黄嘌呤氧化酶抑制剂的设计,合成及生物学评价。
在我们先前的研究中,我们报道了一系列N-苯基异烟酰胺衍生物作为新型黄嘌呤氧化酶(XO)抑制剂,并确定了N-(3-氰基-4-((2-氰基苄氧基)氧基)苯基)异烟酰胺(化合物1)是最有效的一种,IC50值为0.312μM。为了进一步优化结构并提高效能,进行了基于结构的药物设计(SBDD)策略,以构建小分子与XO的Asn768残基之间缺失的H键。我们在苯基的3'位引入一个四唑部分作为H键受体,并获得了一系列N-(3-(1H-四唑-1-基)苯基)异烟酰胺衍生物(2a-t和6-8)。此外,为了研究酰胺逆转的影响,还合成了一些N-(吡啶-4-基)-3-(1H-四唑-1-基)苯甲酰胺衍生物(3c,3e,3i,3k和3u)。并进行评估。生物学评估和结构-活性关系分析表明,3'-(1H-四唑-1-基)部分是N-苯基异烟酰胺骨架的极好片段。为了效力,欢迎在4′-位连接的取代的苄氧基,特别是间-氰基苄氧基(例如2s);而且酰胺的逆转可能会破坏药效,因此维持N-苯基异烟酰胺支架是必不可少的。总而言之,从化合物1开始,SBDD的努力成功鉴定出了有希望的XO抑制剂2s(IC50 = 0.031μM),效力提高了10倍。它的效力非常接近于阳性对照托吡司他(IC50 = 0.021μM)。Lineweaver-Burk图表明化合物2s充当了混合型XO抑制剂。





























 京公网安备 11010802027423号
京公网安备 11010802027423号