当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
In-situ Infrared Spectroscopy Reveals Persistent Alkalinity Near Electrode Surfaces during CO2 Electroreduction
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2019-09-15 , DOI: 10.1021/jacs.9b07000 Kailun Yang 1 , Recep Kas 1 , Wilson A Smith 1
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2019-09-15 , DOI: 10.1021/jacs.9b07000 Kailun Yang 1 , Recep Kas 1 , Wilson A Smith 1
Affiliation
|
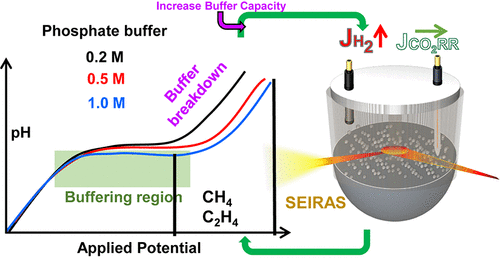
Over the past decade, electrochemical carbon dioxide reduction has become a thriving area of research with the aim of converting electricity to renewable chemicals and fuels. Recent advances through catalyst development have significantly improved selectivity and activity. However, drawing potential dependent structure–activity relationships has been complicated, not only due to the ill-defined and intricate morphological and mesoscopic structure of electrocatalysts, but also by immense concentration gradients existing between the electrode surface and bulk solution. In this work, by using in situ surface enhanced infrared absorption spectroscopy (SEIRAS) and computational modeling, we explicitly show that commonly used strong phosphate buffers cannot sustain the interfacial pH during CO2 electroreduction on copper electrodes at relatively low current densities, <10 mA/cm2. The pH near the electrode surface was observed to be as much as 5 pH units higher compared to bulk solution in 0.2 M phosphate buffer at potentials relevant to the formation of hydrocarbons (−1 V vs RHE), even on smooth polycrystalline copper electrodes. Drastically increasing the buffer capacity did not stand out as a viable solution for the problem as the concurrent production of hydrogen increased dramatically, which resulted in a breakdown of the buffer in a narrow potential range. These unforeseen results imply that most of the studies, if not all, on electrochemical CO2 reduction to hydrocarbons in CO2 saturated aqueous solutions were evaluated under mass transport limitations on copper electrodes. We underscore that the large concentration gradients on electrodes with high local current density (e.g., nanostructured) have important implications on the selectivity, activity, and kinetic analysis, and any attempt to draw structure–activity relationships must rule out mass transport effects.
中文翻译:

原位红外光谱揭示 CO2 电还原过程中电极表面附近的持续碱度
在过去十年中,电化学二氧化碳还原已成为一个蓬勃发展的研究领域,旨在将电力转化为可再生化学品和燃料。通过催化剂开发的最新进展显着提高了选择性和活性。然而,绘制电位相关的结构-活性关系很复杂,这不仅是由于电催化剂的不明确和复杂的形态和介观结构,而且还因为电极表面和本体溶液之间存在巨大的浓度梯度。在这项工作中,通过使用原位表面增强红外吸收光谱 (SEIRAS) 和计算建模,我们明确地表明,在相对较低的电流密度(<10 mA/cm2)下,在铜电极上进行 CO2 电还原期间,常用的强磷酸盐缓冲液无法维持界面 pH 值。在与碳氢化合物形成相关的电位(-1 V vs RHE)下,即使在光滑的多晶铜电极上,电极表面附近的 pH 值也比 0.2 M 磷酸盐缓冲液中的本体溶液高 5 个 pH 单位。大幅增加缓冲容量并不是解决问题的可行方案,因为同时产生的氢气急剧增加,导致缓冲在狭窄的电位范围内崩溃。这些不可预见的结果意味着大多数研究,如果不是全部,在铜电极的传质限制下,评估了在 CO2 饱和水溶液中电化学 CO2 还原为碳氢化合物的效果。我们强调,具有高局部电流密度(例如,纳米结构)的电极上的大浓度梯度对选择性、活性和动力学分析具有重要意义,任何绘制结构-活性关系的尝试都必须排除质量传递效应。
更新日期:2019-09-15
中文翻译:
原位红外光谱揭示 CO2 电还原过程中电极表面附近的持续碱度
在过去十年中,电化学二氧化碳还原已成为一个蓬勃发展的研究领域,旨在将电力转化为可再生化学品和燃料。通过催化剂开发的最新进展显着提高了选择性和活性。然而,绘制电位相关的结构-活性关系很复杂,这不仅是由于电催化剂的不明确和复杂的形态和介观结构,而且还因为电极表面和本体溶液之间存在巨大的浓度梯度。在这项工作中,通过使用原位表面增强红外吸收光谱 (SEIRAS) 和计算建模,我们明确地表明,在相对较低的电流密度(<10 mA/cm2)下,在铜电极上进行 CO2 电还原期间,常用的强磷酸盐缓冲液无法维持界面 pH 值。在与碳氢化合物形成相关的电位(-1 V vs RHE)下,即使在光滑的多晶铜电极上,电极表面附近的 pH 值也比 0.2 M 磷酸盐缓冲液中的本体溶液高 5 个 pH 单位。大幅增加缓冲容量并不是解决问题的可行方案,因为同时产生的氢气急剧增加,导致缓冲在狭窄的电位范围内崩溃。这些不可预见的结果意味着大多数研究,如果不是全部,在铜电极的传质限制下,评估了在 CO2 饱和水溶液中电化学 CO2 还原为碳氢化合物的效果。我们强调,具有高局部电流密度(例如,纳米结构)的电极上的大浓度梯度对选择性、活性和动力学分析具有重要意义,任何绘制结构-活性关系的尝试都必须排除质量传递效应。
































 京公网安备 11010802027423号
京公网安备 11010802027423号