当前位置:
X-MOL 学术
›
J. Am. Chem. Soc.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
CO Coupling Chemistry of a Terminal Mo Carbide: Sequential Addition of Proton, Hydride, and CO Releases Ethenone
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2019-09-03 , DOI: 10.1021/jacs.9b07743 Joshua A Buss 1 , Gwendolyn A Bailey 1 , Julius Oppenheim 1 , David G VanderVelde 1 , William A Goddard 1 , Theodor Agapie 1
Journal of the American Chemical Society ( IF 14.4 ) Pub Date : 2019-09-03 , DOI: 10.1021/jacs.9b07743 Joshua A Buss 1 , Gwendolyn A Bailey 1 , Julius Oppenheim 1 , David G VanderVelde 1 , William A Goddard 1 , Theodor Agapie 1
Affiliation
|
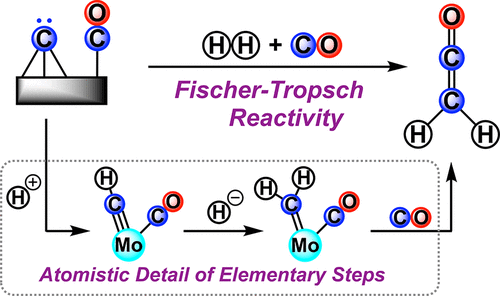
The mechanism originally proposed by Fischer and Tropsch for carbon monoxide (CO) hydrogenative catenation involves C-C coupling from a carbide-derived surface methylidene. A single molecular system capable of capturing these complex chemical steps is hitherto unknown. Herein, we demonstrate the sequential addition of proton and hydride to a terminal Mo carbide derived from CO. The resulting anionic methylidene couples with CO (1 atm.) at low-temperature (-78 °C) to release ethenone. Importantly, the synchronized delivery of two reducing equivalents and an electrophile, in the form of a hydride (H- = 2e- + H+), promotes alkylidene formation from the carbyne precur-sor and enables coupling chemistry, under conditions milder than previous described for single electron reduction steps. Thermodynamic measurements bracket the hydricity and acidity requirements for promoting methylidene formation from carbide as feasible upon heterogenolysis of H2. Methylidene formation prior to C-C coupling proves critical for organic product release, as evidenced by direct carbide carbonylation studies. Spectroscopic studies, a monosilylated model system, and Quantum Mechanics computations provide insight into the mechanistic details of this reaction sequence, which serves as a rare model of the initial stages of the Fischer Tropsch synthesis.
中文翻译:

末端钼碳化物的 CO 偶联化学:连续添加质子、氢化物和 CO 释放乙烯酮
Fischer 和 Tropsch 最初提出的用于一氧化碳 (CO) 氢化链的机制涉及来自碳化物衍生的表面亚甲基的 CC 偶联。能够捕获这些复杂化学步骤的单个分子系统迄今为止是未知的。在此,我们展示了质子和氢化物顺序添加到源自 CO 的末端碳化钼。所得阴离子亚甲基在低温 (-78 °C) 下与 CO (1 atm.) 结合以释放乙酮。重要的是,以氢化物 (H- = 2e- + H+) 的形式同步传递两个还原当量和一个亲电子试剂,可促进碳炔前体形成亚烷基,并在比之前描述的更温和的条件下实现偶联化学单电子还原步骤。热力学测量涵盖了促进从碳化物形成亚甲基的水合度和酸度要求,这在 H2 异质分解时是可行的。直接碳化物羰基化研究证明,在 CC 偶联之前形成亚甲基对有机产物释放至关重要。光谱研究、单甲硅烷基化模型系统和量子力学计算提供了对该反应序列机械细节的洞察,这是费托合成初始阶段的罕见模型。
更新日期:2019-09-03
中文翻译:
末端钼碳化物的 CO 偶联化学:连续添加质子、氢化物和 CO 释放乙烯酮
Fischer 和 Tropsch 最初提出的用于一氧化碳 (CO) 氢化链的机制涉及来自碳化物衍生的表面亚甲基的 CC 偶联。能够捕获这些复杂化学步骤的单个分子系统迄今为止是未知的。在此,我们展示了质子和氢化物顺序添加到源自 CO 的末端碳化钼。所得阴离子亚甲基在低温 (-78 °C) 下与 CO (1 atm.) 结合以释放乙酮。重要的是,以氢化物 (H- = 2e- + H+) 的形式同步传递两个还原当量和一个亲电子试剂,可促进碳炔前体形成亚烷基,并在比之前描述的更温和的条件下实现偶联化学单电子还原步骤。热力学测量涵盖了促进从碳化物形成亚甲基的水合度和酸度要求,这在 H2 异质分解时是可行的。直接碳化物羰基化研究证明,在 CC 偶联之前形成亚甲基对有机产物释放至关重要。光谱研究、单甲硅烷基化模型系统和量子力学计算提供了对该反应序列机械细节的洞察,这是费托合成初始阶段的罕见模型。
































 京公网安备 11010802027423号
京公网安备 11010802027423号