当前位置:
X-MOL 学术
›
J. Chem. Theory Comput.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Coarse-Grained Modeling of the Interplay between Secondary Structure Propensities and Protein Fold Assembly
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-02-27 00:00:00 , DOI: 10.1021/acs.jctc.7b01242 Aleksandra E. Dawid 1 , Dominik Gront 1 , Andrzej Kolinski 1
Journal of Chemical Theory and Computation ( IF 5.7 ) Pub Date : 2018-02-27 00:00:00 , DOI: 10.1021/acs.jctc.7b01242 Aleksandra E. Dawid 1 , Dominik Gront 1 , Andrzej Kolinski 1
Affiliation
|
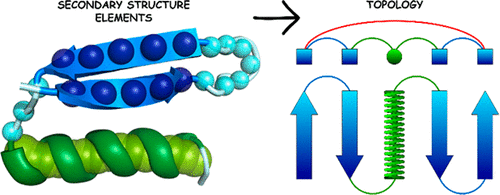
We recently developed a new coarse-grained model of protein structure and dynamics [Dawid et al. J. Chem. Theory Comput. 2017, 13(11), 5766−5779]. The model assumed a single bead representation of amino acid residues, where positions of such united residues were defined by centers of mass of four amino acid fragments. Replica exchange Monte Carlo sampling of the model chains provided good pictures of modeled structures and their dynamics. In its generic form the statistical knowledge-based force field of the model has been dedicated for single-domain globular proteins. Sequence-specific interactions are defined by three-letter secondary structure data. In the present work we demonstrate that different assignments and/or predictions of secondary structures are usually sufficient for enforcing cooperative formation of native-like folds of SURPASS chains for the majority of single-domain globular proteins. Simulations of native-like structure assembly for a representative set of globular proteins have shown that the accuracy of secondary structure data is usually not crucial for model performance, although some specific errors can strongly distort the obtained three-dimensional structures.
中文翻译:

二级结构倾向与蛋白质折叠组装相互作用的粗粒度模型
我们最近开发了一种新的蛋白质结构和动力学的粗粒度模型[Dawid等。J.化学。理论计算。 2017年,13(11),5766-5779]。该模型假定氨基酸残基的单个珠粒表示,其中此类统一残基的位置由四个氨基酸片段的质心定义。模型链的副本交换蒙特卡洛采样提供了建模结构及其动力学的良好图片。以其通用形式,该模型的基于统计知识的力场专用于单域球状蛋白。特定于序列的相互作用是由三个字母的二级结构数据定义的。在目前的工作中,我们证明二级结构的不同赋值和/或预测通常足以对大多数单域球状蛋白强制协作形成SURPASS链的天然样折叠。
更新日期:2018-02-27
中文翻译:
二级结构倾向与蛋白质折叠组装相互作用的粗粒度模型
我们最近开发了一种新的蛋白质结构和动力学的粗粒度模型[Dawid等。J.化学。理论计算。 2017年,13(11),5766-5779]。该模型假定氨基酸残基的单个珠粒表示,其中此类统一残基的位置由四个氨基酸片段的质心定义。模型链的副本交换蒙特卡洛采样提供了建模结构及其动力学的良好图片。以其通用形式,该模型的基于统计知识的力场专用于单域球状蛋白。特定于序列的相互作用是由三个字母的二级结构数据定义的。在目前的工作中,我们证明二级结构的不同赋值和/或预测通常足以对大多数单域球状蛋白强制协作形成SURPASS链的天然样折叠。





























 京公网安备 11010802027423号
京公网安备 11010802027423号