当前位置:
X-MOL 学术
›
J. Phys. Chem. C
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Predicting the Strength of Metal–Support Interaction with Computational Descriptors for Adhesion Energies
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2019-08-08 , DOI: 10.1021/acs.jpcc.9b06893 Elisabeth M. Dietze 1 , Philipp N. Plessow 1
The Journal of Physical Chemistry C ( IF 3.3 ) Pub Date : 2019-08-08 , DOI: 10.1021/acs.jpcc.9b06893 Elisabeth M. Dietze 1 , Philipp N. Plessow 1
Affiliation
|
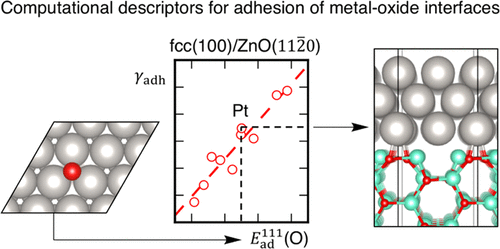
Interfaces play an important role in heterogeneous catalysis where oxides are typically used as supports to stabilize catalytically active transition metal particles and to tune their electronic properties for optimal performance in catalysis. In this contribution, we study numerous metal–oxide interfaces using density functional theory. For a given oxide, variations in adhesion energies with different metals can be described by the adsorption energy of atomic oxygen on the corresponding metal surfaces, thus forming scaling relations similar to those used for adsorbates on metal surfaces. Variations between different oxides can be analyzed through the number of interfacial oxygen atoms that form metal–oxygen bonds. This descriptor can often be derived from the structure of the clean oxide surfaces. Adhesion of the studied interfaces, which is dominated by metal–oxygen bonds, is thus well described with a single scaling relation using two descriptors, one for the metal and one for the oxide surface.
中文翻译:

预测金属与载体间相互作用力的强度,并使用计算描述符进行黏合能
界面在非均相催化中起着重要作用,其中氧化物通常用作支撑物,以稳定催化活性过渡金属颗粒并调整其电子性能,以实现最佳催化性能。在这项贡献中,我们使用密度泛函理论研究了许多金属-氧化物界面。对于给定的氧化物,可以通过原子氧在相应金属表面上的吸附能来描述与不同金属的粘附能的变化,从而形成与用于金属表面上的吸附物相似的结垢关系。可以通过形成金属-氧键的界面氧原子数来分析不同氧化物之间的差异。该描述符通常可以从干净的氧化物表面的结构中得出。所研究界面的粘合性,
更新日期:2019-08-09
中文翻译:
预测金属与载体间相互作用力的强度,并使用计算描述符进行黏合能
界面在非均相催化中起着重要作用,其中氧化物通常用作支撑物,以稳定催化活性过渡金属颗粒并调整其电子性能,以实现最佳催化性能。在这项贡献中,我们使用密度泛函理论研究了许多金属-氧化物界面。对于给定的氧化物,可以通过原子氧在相应金属表面上的吸附能来描述与不同金属的粘附能的变化,从而形成与用于金属表面上的吸附物相似的结垢关系。可以通过形成金属-氧键的界面氧原子数来分析不同氧化物之间的差异。该描述符通常可以从干净的氧化物表面的结构中得出。所研究界面的粘合性,














































 京公网安备 11010802027423号
京公网安备 11010802027423号