当前位置:
X-MOL 学术
›
Nat. Commun.
›
论文详情
Our official English website, www.x-mol.net, welcomes your
feedback! (Note: you will need to create a separate account there.)
Mendelian randomization integrating GWAS and eQTL data reveals genetic determinants of complex and clinical traits.
Nature Communications ( IF 14.7 ) Pub Date : 2019-07-24 , DOI: 10.1038/s41467-019-10936-0 Eleonora Porcu 1, 2 , Sina Rüeger 2, 3 , Kaido Lepik 3, 4 , , , Federico A Santoni 5 , Alexandre Reymond 1 , Zoltán Kutalik 2, 3
Nature Communications ( IF 14.7 ) Pub Date : 2019-07-24 , DOI: 10.1038/s41467-019-10936-0 Eleonora Porcu 1, 2 , Sina Rüeger 2, 3 , Kaido Lepik 3, 4 , , , Federico A Santoni 5 , Alexandre Reymond 1 , Zoltán Kutalik 2, 3
Affiliation
|
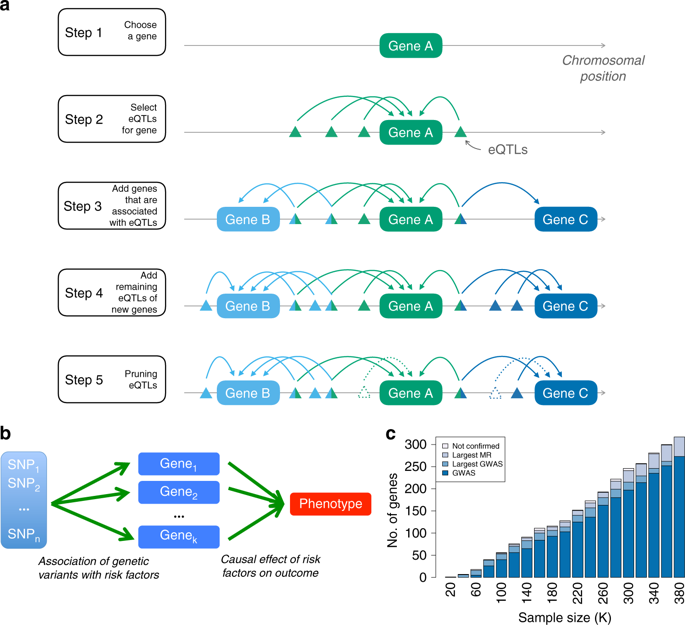
Genome-wide association studies (GWAS) have identified thousands of variants associated with complex traits, but their biological interpretation often remains unclear. Most of these variants overlap with expression QTLs, indicating their potential involvement in regulation of gene expression. Here, we propose a transcriptome-wide summary statistics-based Mendelian Randomization approach (TWMR) that uses multiple SNPs as instruments and multiple gene expression traits as exposures, simultaneously. Applied to 43 human phenotypes, it uncovers 3,913 putatively causal gene-trait associations, 36% of which have no genome-wide significant SNP nearby in previous GWAS. Using independent association summary statistics, we find that the majority of these loci were missed by GWAS due to power issues. Noteworthy among these links is educational attainment-associated BSCL2, known to carry mutations leading to a Mendelian form of encephalopathy. We also find pleiotropic causal effects suggestive of mechanistic connections. TWMR better accounts for pleiotropy and has the potential to identify biological mechanisms underlying complex traits.
中文翻译:

整合 GWAS 和 eQTL 数据的孟德尔随机化揭示了复杂和临床特征的遗传决定因素。
全基因组关联研究 (GWAS) 已经确定了数千种与复杂性状相关的变异,但它们的生物学解释通常仍不清楚。大多数这些变体与表达 QTL 重叠,表明它们可能参与基因表达的调控。在这里,我们提出了一种基于转录组范围汇总统计的孟德尔随机化方法 (TWMR),该方法使用多个 SNP 作为工具,同时使用多个基因表达特征作为暴露。应用于 43 种人类表型,它揭示了 3,913 个假定的因果基因-性状关联,其中 36% 在之前的 GWAS 中没有附近的全基因组显着 SNP。使用独立的关联汇总统计,我们发现这些位点中的大多数由于功率问题而被 GWAS 遗漏。在这些联系中值得注意的是与教育程度相关的 BSCL2,已知其携带导致孟德尔形式脑病的突变。我们还发现多效因果效应暗示了机械联系。TWMR 更好地解释了多效性,并有可能识别复杂性状背后的生物学机制。
更新日期:2019-07-24
中文翻译:
整合 GWAS 和 eQTL 数据的孟德尔随机化揭示了复杂和临床特征的遗传决定因素。
全基因组关联研究 (GWAS) 已经确定了数千种与复杂性状相关的变异,但它们的生物学解释通常仍不清楚。大多数这些变体与表达 QTL 重叠,表明它们可能参与基因表达的调控。在这里,我们提出了一种基于转录组范围汇总统计的孟德尔随机化方法 (TWMR),该方法使用多个 SNP 作为工具,同时使用多个基因表达特征作为暴露。应用于 43 种人类表型,它揭示了 3,913 个假定的因果基因-性状关联,其中 36% 在之前的 GWAS 中没有附近的全基因组显着 SNP。使用独立的关联汇总统计,我们发现这些位点中的大多数由于功率问题而被 GWAS 遗漏。在这些联系中值得注意的是与教育程度相关的 BSCL2,已知其携带导致孟德尔形式脑病的突变。我们还发现多效因果效应暗示了机械联系。TWMR 更好地解释了多效性,并有可能识别复杂性状背后的生物学机制。
































 京公网安备 11010802027423号
京公网安备 11010802027423号